



Sporadic fatal insomnia (SFI) is a very rare condition sharing clinical features with the familial form. It presents with highly heterogeneous symptoms, and is characterised by rapid progression and a short life expectancy. An anatomical pathology study is needed for definitive diagnosis; the D178N mutation in the prion protein gene must also be ruled out.1,2 We present the case of a patient with a phenotype suggestive of progressive supranuclear palsy (PSP) of rapid progression.
Clinical caseOur patient was a 50-year-old man whose father had died at an old age due to probable Lewy body dementia; the patient had no other relevant family history. The only relevant personal history was syphilis with cutaneous manifestations, which was treated with penicillin.
The patient initially presented neuropsychiatric symptoms: depression, social isolation, and feelings of disability, which had a negative impact on his personal and work life. His initial contact with the neurology department was due to insidious onset of binocular diplopia. Over the following months, the condition progressed rapidly, with motor slowing associated with impaired manipulative dexterity, instability with frequent falls, and speech alterations associated with voice changes. Our patient’s family noticed significant personality changes, and he himself reported memory and concentration problems. During clinical history taking, the only dysautonomic symptom reported by our patient was erectile dysfunction. He did not have any obvious sleep disorder.
We present the results of the physical examination performed approximately 10 months after symptom onset. General examination detected no pathological signs. From a neuropsychological viewpoint, the patient presented alterations in executive function and memory, but no apraxia or visuospatial alterations; he also displayed frontal release signs (Montreal Cognitive Assessment score: 23/30). Language was fluent, with no signs of dysphasia, although he presented scanning speech and dysprosody. The ophthalmological examination detected limited vertical gaze, impaired eye tracking, and square wave jerks. Our patient also presented generalised bradykinesia with predominantly axial rigidity, without tremor, myoclonus, fasciculations, or trophic changes. Muscle strength was normal. Stretch reflexes were hyperactive in all muscle groups, with bilateral Hoffman sign and flexor plantar reflex. Superficial and deep-tissue sensitivity was normal. Coordination and gait tests revealed dysmetria of all 4 limbs, wide-based stance, and unsteady gait.
Our patient presented a rapidly progressive multisystem neurological disorder. The main differential diagnoses were neurodegenerative, dysimmune, and prion diseases. PSP was the main suspected diagnosis at symptom onset. Among immune-mediated diseases, encephalitis with antibodies against IgLON5, DPPX, mGluR1, or GAD65 was considered the most likely hypothesis. Other possible diagnoses were neurosyphilis, Niemann–Pick disease type C, Whipple disease, and hereditary degenerative ataxias.
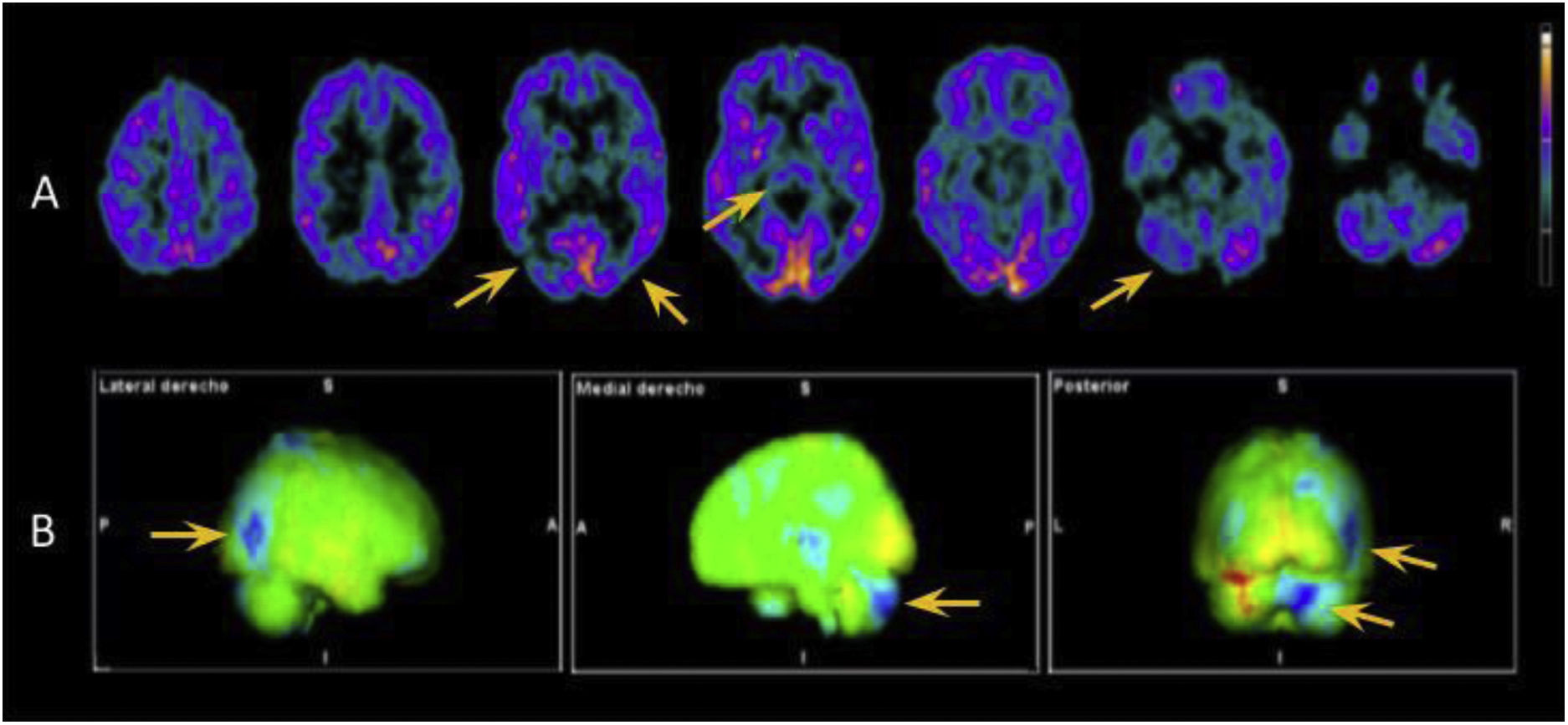
In view of the wide range of potential diagnoses, we conducted extensive complementary testing. Brain MRI detected no relevant alterations. CSF analysis detected elevated protein levels (66 mg/dL; normal range: < 45 mg/dL), without pleocytosis or glucose uptake; a microbiological study (including a VDRL test and PCR for Tropheryma whipplei) yielded negative results and a cytological analysis detected no abnormalities. A CSF and blood autoimmunity study, including antibodies against IgLON5, CASPR2, DPPX, mGluR1, GAD65, AMPAr, NMDAr, GABAr, and LGI1, yielded negative results. We did detect increased levels of total tau protein at 467 pg/mL (pathological range for the patient’s age: > 116 pg/mL), with normal levels of beta-amyloid and phosphorylated tau protein. CSF analysis did not detect 14-3-3 protein, and CSF real-time quaking-induced conversion (RT-QuIC) did not detect prion protein. Several awake EEG studies were performed, revealing no pathological patterns. However, 18F-FDG PET (Fig. 1) and 123I-ioflupane SPECT studies did detect pathological findings (reduced uptake in both striatal nuclei).
 Axial images showing hypometabolism in the thalamus, predominantly right temporo-parieto-occipital junction, and right cerebellar hemisphere (yellow arrows). B) 3D-stereotactic surface projection maps comparing these findings against data from healthy patients of the same age group, which confirm our conclusions. Areas of hypometabolism are indicated in blue.")
18F-FDG PET study. A) Axial images showing hypometabolism in the thalamus, predominantly right temporo-parieto-occipital junction, and right cerebellar hemisphere (yellow arrows). B) 3D-stereotactic surface projection maps comparing these findings against data from healthy patients of the same age group, which confirm our conclusions. Areas of hypometabolism are indicated in blue.
During follow-up, we trialled different treatments, including high-dose corticosteroids, intravenous immunoglobulins, and levodopa (starting dose of 250 mg and maintenance dose of 100 mg every 8 h), without success. Our patient’s clinical status continued to worsen, with more severe cognitive impairment, severe ataxia with inability to walk independently, and severe dysphagia. Approximately a year and a half after symptom onset, he died due to a respiratory infection.
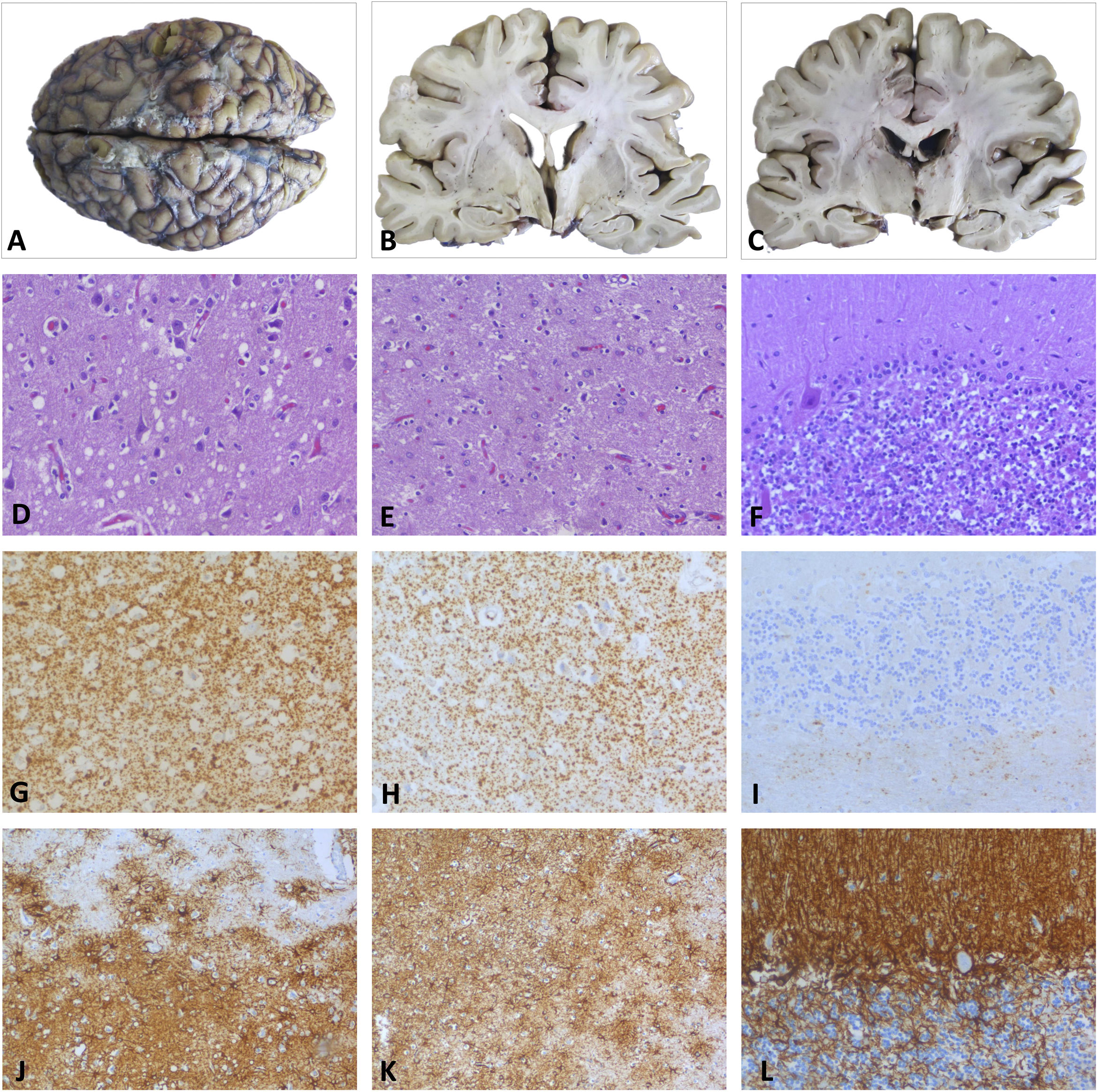
DiagnosisA post mortem anatomopathological examination revealed neurodegenerative changes typical of fatal insomnia, characterised by atrophy and reactive astrocytic gliosis in the thalamus and inferior olivary nuclei, as well as spongiform changes with small vacuoles in cortical regions and diffuse synaptic deposition of prion protein. In the cerebellum, abnormalities mainly involved the vermis and right cerebellar cortex, with moderate cell loss and diffuse foci of prion protein deposition and gliosis (Fig. 2).3,4 Genetic and molecular analyses ruled out the D178N mutation in the prion protein gene and found the MM2 form at codon 129, which led to the definitive diagnosis of SFI.5–10
 Macroscopic study. Images of the whole brain and coronal sections at the level of basal ganglia and thalamus, revealing slight atrophy of the temporal cortex, without other significant changes. D, G, J) Parietal cortex. Neuronal loss and moderate neuropil spongiosis, with small-/medium-sized rounded vacuoles observed with HE staining (D). The immunohistochemical study revealed PrP deposition with a diffuse granular-synaptic pattern (G), as well as marked reactive astrogliosis detected by GFAP staining (J). E, H, K) Thalamus. HE (E) and GFAP staining (K) revealed neuronal loss with minimal spongiosis and severe astrogliosis. Prp deposition in the thalamus was less marked than in the cortex (H). F, I, L) Cerebellum. Moderate loss of granule neurons and Purkinje cells observed with HE staining (F), with small foci of PrP in the granular layer (I) and astrogliosis detected with GFAP (L). GFAP: glial fibrillary acidic protein; HE: haematoxylin-eosin; PrP: prion protein.")
Post mortem histopathological examination. A–C) Macroscopic study. Images of the whole brain and coronal sections at the level of basal ganglia and thalamus, revealing slight atrophy of the temporal cortex, without other significant changes. D, G, J) Parietal cortex. Neuronal loss and moderate neuropil spongiosis, with small-/medium-sized rounded vacuoles observed with HE staining (D). The immunohistochemical study revealed PrP deposition with a diffuse granular-synaptic pattern (G), as well as marked reactive astrogliosis detected by GFAP staining (J). E, H, K) Thalamus. HE (E) and GFAP staining (K) revealed neuronal loss with minimal spongiosis and severe astrogliosis. Prp deposition in the thalamus was less marked than in the cortex (H). F, I, L) Cerebellum. Moderate loss of granule neurons and Purkinje cells observed with HE staining (F), with small foci of PrP in the granular layer (I) and astrogliosis detected with GFAP (L).
GFAP: glial fibrillary acidic protein; HE: haematoxylin-eosin; PrP: prion protein.
We present a case of SFI in which diagnosis was based on conclusive anatomical pathology, genetic, and molecular findings. The disease manifested mainly with cognitive and motor symptoms, with a PSP-like phenotype associated with severe ataxia and atypical progression, without clear sleep disturbances. This, together with the negative results for 14-3-3 and prion protein (RT-QuIC) hindered ante mortem suspicion of a prion disease such as fatal insomnia.11–13 Several reviews have reported similar findings from CSF tests for 14-3-3, RT-QuIC, and tau protein, which differentiate fatal insomnia from other more frequent prion diseases.2,14 Furthermore, results from nuclear medicine brain scans were more relevant than those of brain MRI. Thalamic hypometabolism is a noteworthy finding that may help raise suspicion of fatal insomnia in patients with compatible symptoms.12,15 Our patient did not undergo a polysomnography study due to the lack of specific symptoms; however, this test may have provided useful data for diagnosis.2,8
Fatal insomnia (both sporadic and familial) represents a clinical challenge due to its rareness and the non-specific findings of complementary tests. The condition should be considered in patients presenting a rapidly progressive multisystem neurological disorder, with negative complementary test results. In these cases, brain 18F-FDG PET may guide diagnosis. We wish to stress the importance of post mortem anatomical pathology studies in inconclusive, complex neurological cases.
Please cite this article as: Romero-Fábrega JC, Lorenzo-López R, Rivas-Infante E, Escamilla-Sevilla F, Rashki M, Mínguez-Castellanos A, et al. Insomnio fatal esporádico: fenotipo PSP-like rápidamente progresivo. Neurología. 2024. https://doi.org/10.1016/j.nrl.2023.05.004
