


La displasia tanatofórica (DT) es la osteocondrodisplasia más letal que existe en el periodo neonatal. Se caracteriza por miembros cortos, macrocefalia, tronco de longitud normal y tórax estrecho con hipoplasia pulmonar. Se clasifica en dos subtipos clínicamente definidos: la DT tipo I (DTI), es el subtipo más común y se caracteriza por presentar huesos largos curvados (fémur en forma de receptor telefónico) con o sin deformaciones craneales, y la DT tipo II (DTII), en que los fetos presentan cráneos en forma de “hoja de trébol” y fémures rectos. Muchas de las alteraciones morfológicas de este tipo de enanismo coinciden en ambos subtipos: micrognatia o platispondilia vertebral. Actualmente existen varias formas de diagnosticar este trastorno por imágenes clínicas o técnicas moleculares. Se presentan los hallazgos clínicos y de imagen de un feto de 36 semanas de gestación que presentó macrocefalia, frente prominente, puente nasal plano, costillas cortas y gruesas, tórax estrecho, micromelia extrema y vértebras planas, asociados a una rotura prematura de membrana, en que se estableció el diagnóstico de DT tipo II.
Thanatophoric dysplasia (TD) is the most lethal osteochondrodysplasia disorder in the neonatal period. Characteristics of TD include severe shortening of the limbs, macrocephaly, normal trunk length and narrow thorax with pulmonary hypoplasia. It is classified into 2 clinically defined subtypes: TD type I (TDI) is the most common subtype, characterised by curved long bones (shaped like a telephone receiver) with or without skull deformities; and TD type II, in which the foetus has a cloverleaf-shaped skull and straight femurs. In this type of dwarfism most of the morphological alterations, like micrognathia and platyspondyly of the vertebrae, coincide in both types. Currently, the diagnosis of this disorder can be performed by clinical imaging and molecular techniques. The case report shares the clinical and sonographic findings of a 36-week-old foetus that presented macrocephaly, low nasal bridge, short ribs, narrow thorax, marked shortening of the limbs (micromelia) and platyspondyly of the vertebrae, associated with premature membrane rupture. A diagnosis of TD type II was established.
La displasia tanatofórica (DT) es la más frecuente de las condrodisplasias incompatibles con la vida en fetos y neonatos. El término tanatofórica deriva del griego y significa “llevar a la muerte”. Fue descrita por primera en 1967 por Maroteaux y cols.1. Tiene una prevalencia que varía de 0.21 a 0.30 por cada 10,000 nacidos vivos (1/33,330 a 1/47,620)2. Se caracteriza por presentar un acortamiento marcado de las extremidades con numerosos repliegues de piel, tronco de tamaño relativamente normal, tórax angosto (en campana), cabeza desproporcionadamente grande, prominencia frontal, protrusión ocular, puente nasal hundido, y con menor frecuencia cráneo en “hoja de trébol”3,4.
Se distinguen dos tipos de DT. En el tipo I los huesos tubulares, particularmente el fémur, adoptan una forma curvada (forma de receptor telefónico) y los cuerpos vertebrales se aplanan. En el tipo II, los huesos del fémur son rectos, los cuerpos vertebrales no son tan planos como en la DTI y la craneosinostosis es muy común (>90%), que se acompaña de cráneo en “hoja de trébol” en más del 50% de los casos. En contraste, la DTI se asocia con menor frecuencia a craneosinostosis (<30%), con leves rasgos de cráneo en “hoja de trébol” (3%)5. Todas estas características facilitan su diagnóstico radiológico y ultrasonográfico bidimensional.
La herencia es autosómica dominante. En la DTI, la mutación más frecuente se encuentra a nivel de la región de enlace entre IgII e IgIII: 742C r T, lo que se traduce en Arg248Cys. En la DTII, la causa es una mutación de la segunda cinasa a nivel del receptor 3 del factor de crecimiento de fibroblastos (FGFR3) 1948A r G, lo que se traduce en Lys650Glu6.
PatogeniaDistintas mutaciones del gen FGFR3 del cromosoma 4 son la causa de tres displasias esqueléticas de miembros cortos: DT, acondroplasia e hipocondroplasia. Si estos trastornos se organizaran según su gravedad, del más leve al más grave, su orden sería: hipocondroplasia, acondroplasia y DT7. La DT expresa las malformaciones más graves de todas las osteocondrodisplasias8.
Las transcripciones del gen FGFR3 se expresan principalmente en cartílago y en el sistema nervioso central. La función del FGFR3 puede deducirse del Fgfr3 del ratón knockout, el cual desarrolla fémur y vértebras excesivamente largas, lo que da como resultado una cola enorme. Por ende, la función normal del FGFR3 es regular la osificación endocondral. Las mutaciones del FGFR3 en la acondroplasia y la DT muestran un nivel elevado de fosfotirosina. Las señales constitutivas dependientes de ligando que se producen en estas mutaciones conllevan la maduración prematura de los huesos craneales y de todo el esqueleto9.
Las mutaciones del FGFR3 en displasias esqueléticas de miembros cortos, como son hipocondroplasia, acondroplasia y DT, son potenciadoras de su función normal, que consiste en regular la osificación endocondral6.
Diagnóstico ultrasonográficoLa medición ecográfica de la longitud femoral, en especial cuando se relaciona con el diámetro biparietal, constituye un método fiable para la identificación de ciertas formas de displasias esqueléticas con acortamiento de miembros, incluyendo la DT. El diagnóstico ecosonográfico se puede sospechar sobre la base de un enanismo de miembros cortos asociado con hipoplasia pulmonar grave10. La DTI se caracteriza por fémur y húmeros rizomélicos y curvos con metáfisis desgastadas. Las manos y los pies tienen un tamaño y aspecto normal, pero los dedos son cortos y poseen una forma de salchicha. Las costillas son muy cortas, y en el plano mediosagital puede observarse la hipoplasia torácica grave a nivel de la unión toracoabdominal. La cabeza es grande, con protrusión de los huesos frontales e hipoplasia o ausencia del hueso nasal. El cráneo tiene una base corta y con frecuencia se encuentra una disminución del diámetro del foramen magno11. En la DTII se reconoce la típica “cabeza en trébol”, en el plano coronal de la cabeza fetal, que es causada por una sinostosis de las suturas lambdoidea, coronal y sagital, que conlleva la protrusión de los temporales. Los fémures son cortos, pero no tan cortos como en la DTI; estos tienden a ser más rectos. A pesar de estos hallazgos, el tronco tiene una longitud normal. La columna vertebral muestra cuerpos vertebrales aplanados con espacios vertebrales ensanchados, lo que da una apariencia radiológica de “H”. Otros hallazgos asociados pueden ser la presencia de tejido blando redundante, polihidramnios, riñones en herradura, defectos del septo auricular, válvula tricúspide defectuosa, ano imperforado y sinostosis radioulnar12.
Reporte de casoMujer de 35 años de edad que fue atendida en la sala de emergencia del Hospital Escuela Bertha Calderón Roque (HEBCR) de Managua, Nicaragua, por la presencia de salida de líquido transvaginal claro, inodoro y de color amarillento. Fue ingresada a la sala de Alto Riesgo Obstétrico con el siguiente diagnóstico: multigesta con 2 partos anteriores y ningún aborto, embarazo de 36 semanas de gestación (SG) por fecha de última menstruación, ruptura prematura de membranas de 2½ horas de evolución, feto en presentación pélvica y paridad satisfecha. La paciente no presentó antecedentes personales ni familiares patológicos relevantes; no se encontraron antecedentes de malformaciones en los productos de las gestaciones previas. No había antecedentes de consanguinidad o anomalías esqueléticas en su familia. Era una mujer dedicada al hogar, con educación secundaria. Negó ingesta de tóxicos o medicamentos contraindicados durante el embarazo.
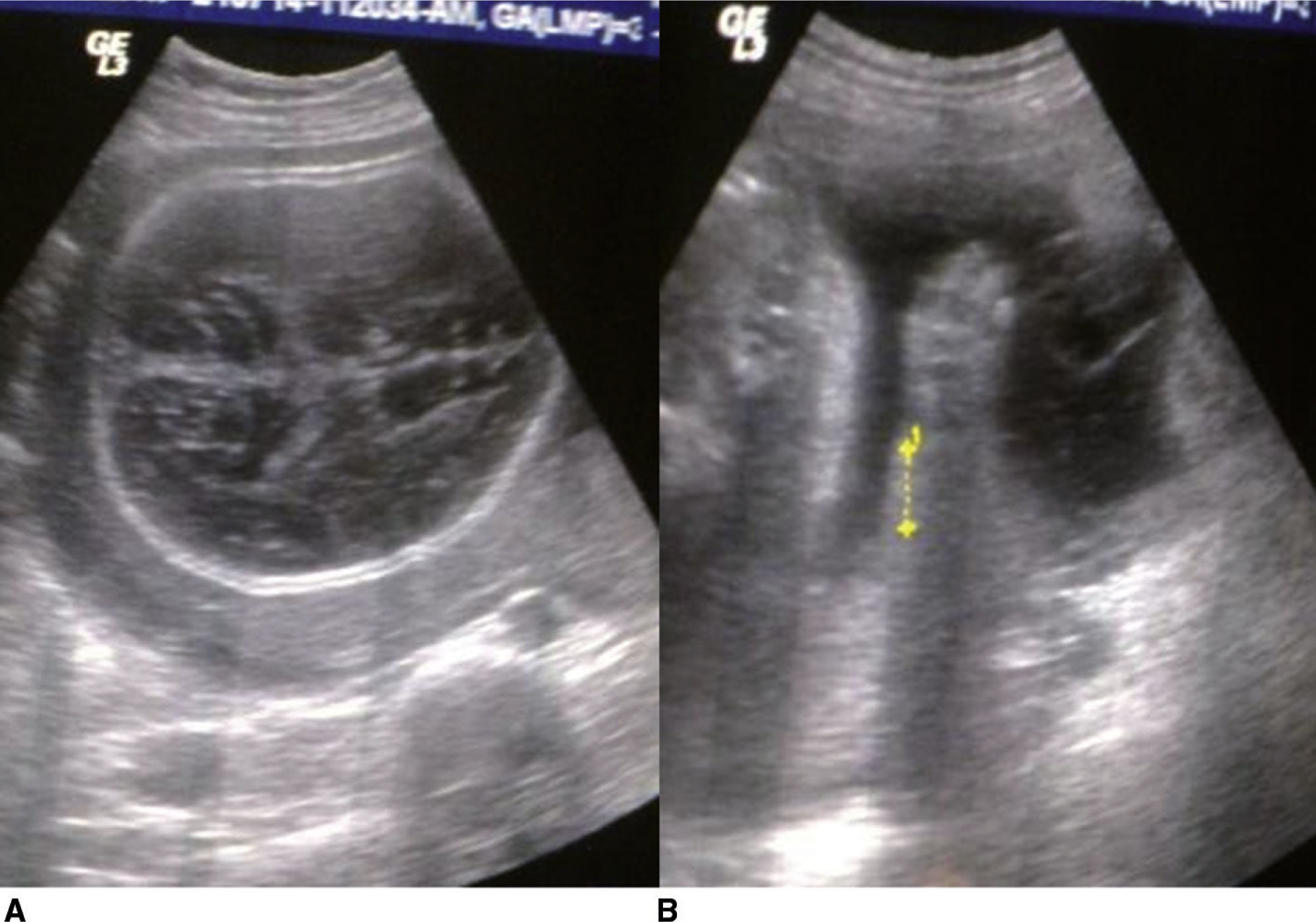
Se realizaron cuatro exámenes ultrasonográficos, los cuales reportaron un producto con anomalías morfológicas. El primer ultrasonido reveló un embarazo de 204/7 SG, más ventriculomegalia bilateral de los ventrículos laterales y fémur corto. El último ultrasonido mostró un feto de 27 SG en presentación cefálica, situación longitudinal y posición en oblicuo izquierdo. Se encontró asimetría biométrica del feto, con: diámetro biparietal = 85mm (34.3 SG); circunferencia cefálica = 287mm (31.5 SG); circunferencia abdominal = 256mm (26.4 SG); longitud femoral = 26mm (28.5 SG), y peso fetal estimado = 665 g. A nivel craneal se observó cráneo en trébol y datos de deformación a la irrigación con el transductor, con ambos tálamos presentes y fosa posterior normal. Ausencia de hueso nasal, asociado a datos de micrognatia. Deformidad a nivel de columna vertebral, con datos de hiperecogenicidad en arcos costales que impresionan imagen en rosario. Huesos tubulares de miembros superiores e inferiores acortados (6 semanas menor con relación a abdomen). Falanges proximales y distales de extremidades superiores e inferiores hipoplásicas (figs. 1 A y B).
. Huesos largos 6 semanas más cortos en relación con la edad gestacional (B). Micromelia de miembros.")
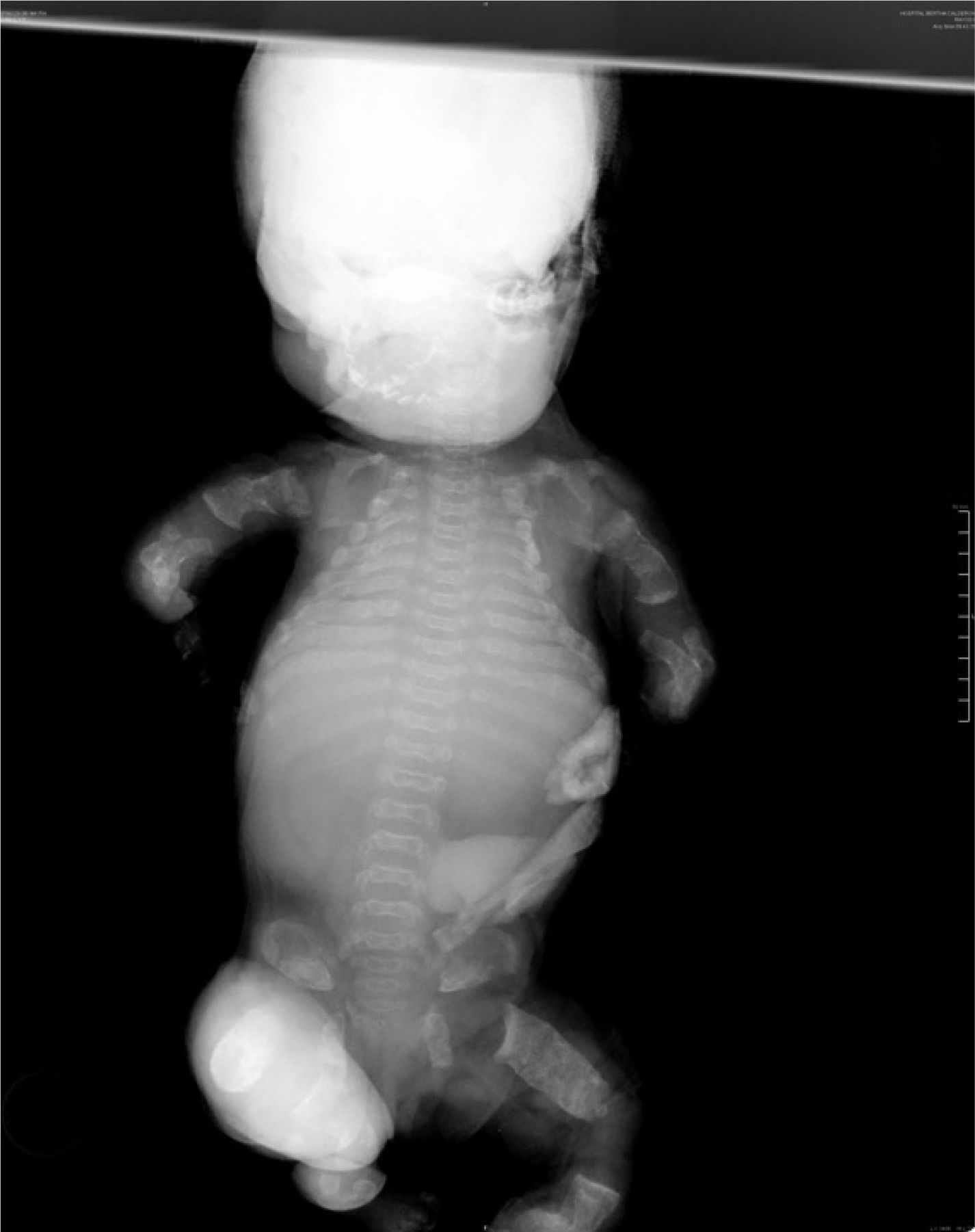
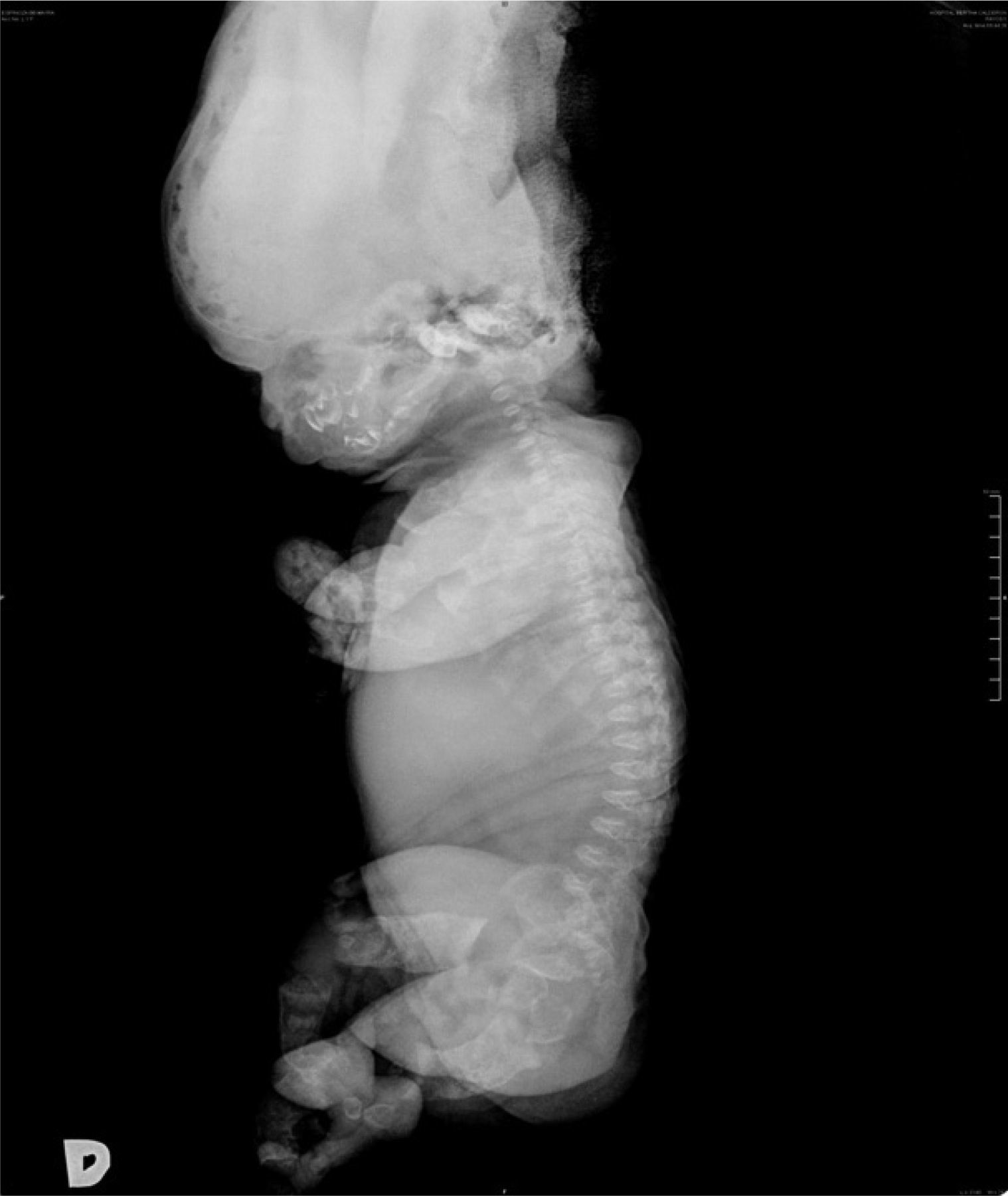
Al nacimiento, el examen físico del neonato reveló macrocefalia de aspecto anormal por fractura, frente prominente, puente nasal hundido, tórax estrecho con importante dificultad respiratoria, abdomen prominente y micromelia extrema. El sexo era femenino y los estudios radiológicos destacaron: bóveda craneal de aspecto anómalo y base pequeña; tórax estrecho, clavículas en posición elevada, costillas cortas con ensanchamiento de los extremos anteriores y clavículas pequeñas; platispondilia marcada; pelvis hipoplásica, extremidades con huesos largos acortados y diáfisis rectas; huesos de manos y pies anchos y cortos (figs. 2 y 3).
Las conclusiones diagnósticas fueron:
- •
Embarazo de 27 semanas.
- •
DTII, con un pronóstico fetal malo para vida y función.
El diagnóstico diferencial incluyó otras displasias esqueléticas que se caracterizan por micromelia e hipoplasia torácica grave, como acondrogénesis, hipofosfatemia y osteogénesis imperfecta tipo II. En la acondrogénesis se pueden encontrar otros hallazgos como la micrognatia e hipomineralización importante, que están ausentes en la DT13. La hipofosfatemia se caracteriza, además de micromelia e hipoplasia torácica, por hipomineralización. Las fracturas solamente se observan en la osteogénesis imperfecta letal grado II. Con respecto a la DTII, el cráneo en hoja de trébol puede encontrarse en síndromes raros como el de Pfeiffer (anomalías esqueléticas distintas) y el síndrome de Crouzon, en que no hay acortamiento de miembros. También se puede incluir dentro del diagnóstico diferencial al síndrome de Ellis van Creveld, la displasia asfixiante de tórax y el síndrome de acortamiento costal con polidactilia10.
DiscusiónLa DT ocurre en uno de cada 20,000 nacimientos y muestra predominio en el género masculino sobre el femenino en una proporción de 2:113. Se debe a mutaciones del gen del brazo corto del cromosoma 4 que codifica el receptor 3 del factor de crecimiento fibroblástico. Se han descrito varias mutaciones sin sentido en el gen de este receptor, que permiten dividir la DT en los tipos I y II; esta última, llamada también DT con cráneo treboliforme, no tiene la curvatura clásica del fémur que se describe en la DTI. Dado que la mutación adicionalmente dispara la actividad de la tirosina cinasa del receptor, y como normalmente el FGFR3 es un regulador negativo de crecimiento, la mutación permite al receptor activado enviar señales negativas dentro de los condrocitos, lo que lleva a una desorganización generalizada de la osificación endocondral en el plato epifisiario fetal7.
La probabilidad de recurrencia en el padecimiento se ha documentado entre el 2% y el 12%, tomando en cuenta los casos explicados por mosaicismo germinal en alguno de los padres14. Se han reportado casos de DT relacionados con exposición materna a infecciones, radiaciones y tratamiento antiviral15. En ciertos casos, esta anomalía congénita se asocia con polihidramnios16.
La causa de la muerte en las DT habitualmente ocurre por falla cardiorrespiratoria secundaria a hipoplasia pulmonar condicionada por una caja torácica estrecha, lo cual ocurrió en nuestro caso a los poco minutos del nacimiento. Las herramientas de estudio prenatal actuales permiten diagnosticar las DT en diferentes momentos del embarazo. Si bien la ecografía permite medir los huesos largos desde la décima semana de gestación y el padecimiento se ha llegado a sospechar desde la semana 15 de la gestación, observar las porciones osificadas del esqueleto fetal y los encorvamientos de las extremidades solo es posible a finales del segundo trimestre, de tal manera que el diagnóstico de las displasias esqueléticas letales es posible a la semana 24.
En este caso se realizó una identificación temprana de los signos por imagen de la enfermedad (fig. 1). La identificación de huesos cortos durante el segundo trimestre hace sospechar una anormalidad muscoloesquelética fetal. Un acortamiento discreto del fémur o del húmero puede ser la expresión de una aberración cromosómica, de un retraso armónico en el crecimiento intrauterino o de una displasia esquelética. Hay que tener en cuenta que la longitud femoral tiene una relación directa con el crecimiento de otros marcadores biométricos. La longitud del pie fetal es aproximadamente igual a la longitud del fémur durante todo el curso del embarazo. Este parámetro puede ser de utilidad para la detección de una displasia esquelética. La relación longitud femoral/longitud del pie debe ser de 1.
Los criterios radiológicos característicos de la DT, presentes en nuestro caso, fueron: aplanamiento importante de los cuerpos vertebrales (platispondilia) que adoptan la forma de letra ”H” o”U”invertida, aumento de los espacios intervertebrales y acortamiento importante de los huesos largos de las cuatro extremidades (figs. 2 y 3).
.")

Los criterios morfológicos que caracterizan a la DT incluyen: cabeza grande con prominencia frontal, depresión nasal del puente, tronco relativamente normal, tórax estrecho en forma de campana e importante acortamiento rizomélico de las 4 extremidades, lo cual se cree que es debido a la actividad mitótica reducida en las células cartilaginosas de la zona de proliferación de crecimiento endocondral de los huesos largos y pulmones hipoplásicos, a causa de la estrechez de la cavidad torácica. Todas estas características estuvieron presentes en el caso que se comenta. El estudio molecular permite tener la confirmación diagnóstica desde etapas tan tempranas como la semana 10 al obtener células para análisis de ácido desoxirribonucleico por biopsia de vellosidades coriales.
El control prenatal es de suma importancia, ya que cuanto más temprano se haga el diagnóstico de esta enfermedad el pronóstico para la madre mejora, puede iniciarse el consejo genético y darle a los padres apoyo multidisciplinario.