





La esclerosis lateral amiotrófica es una enfermedad neurodegenerativa progresiva crónica de etiología desconocida que afecta de forma selectiva a las motoneuronas tanto superior como inferior. Hoy en día, su diagnóstico se basa en hallazgos clínicos, electromiográficos y la exclusión de otras enfermedades con síntomas similares, no existiendo pruebas específicas ni marcadores biológicos que confirmen su diagnóstico.
En este artículo exponemos las nuevas técnicas de RM como la transferencia de magnetización, la espectroscopia por RM, la RM funcional, el tensor difusor o las técnicas volumétricas, que pueden contribuir tanto a la confirmación diagnóstica precoz como al seguimiento de esta enfermedad. Revisamos la utilidad de dichas técnicas según la bibliografía y discutimos sus posibles aplicaciones en la práctica clínica.
Amyotrophic lateral sclerosis is a chronic progressive neurodegenerative disease of unknown etiology. It selectively affects both upper and lower motor neurons. Nowadays, amyotrophic lateral sclerosis is diagnosed on the basis of clinical and electromyographic findings and by ruling out other diseases with similar symptoms. To date, there are no specific tests or biologic markers to confirm the diagnosis.
This article discusses new MRI techniques like magnetization transfer imaging, MR spectroscopy, functional MRI, diffusion tensor imaging, and volumetric techniques that can contribute to early confirmation of the diagnosis and to the follow-up of this disease. We review the usefulness of each of these techniques and discuss their possible applications in clinical practice.
La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa poco frecuente, difícil de diagnosticar en sus inicios, y lamentablemente de curso rápidamente progresivo, para la cual no existe en la actualidad un tratamiento eficaz.
Puesto que no existen pruebas específicas para el diagnóstico de ELA y debido a que su forma de presentación clínica puede simular muchas otras enfermedades, su diagnóstico a menudo se ve retrasado, con las consiguientes consecuencias sobre la aplicación de un tratamiento precoz, que pueda prolongar la supervivencia de estos pacientes o al menos mejorar su calidad de vida. Por esta razón son muchos los trabajos de investigación que se están llevando a cabo, con el fin de intentar conseguir una técnica de imagen que permita un diagnóstico temprano de la enfermedad. Las técnicas convencionales de RM son poco específicas y únicamente son útiles para descartar otros procesos que puedan simular una ELA. Las nuevas técnicas de RM tienen un papel esperanzador en el diagnóstico de la afectación de la primera motoneurona, y junto con la valoración clínica y electromiográfica están permitiendo obtener un diagnóstico de confirmación en estadios más precoces de la enfermedad.
DiscusiónLa ELA, o enfermedad de Lou Gehrig en honor al famoso jugador de béisbol que la presentó, es una grave enfermedad neurológica crónica degenerativa que se caracteriza por la afectación selectiva de las neuronas motoras, tanto superior como inferior1. Su incidencia mundial varía de 0,5-2 sujetos por cada 100.000 y su prevalencia es de 5/100.000 personas1. La forma hereditaria constituye tan solo un 10% de los casos aproximadamente, mientras que el gran porcentaje de los casos continúa siendo de etiología desconocida2. Se han recogido más de 100 mutaciones distintas, la mayoría autosómicas dominantes con penetrancia variable en la ELA familiar1,2.
La incidencia es discretamente mayor en hombres que en mujeres con un cociente de 2/1, presentándose con mayor frecuencia en un rango de edad de 40 a 60 años y con una supervivencia media de 3 a 5 años desde su instauración1,3,4. Las complicaciones respiratorias constituyen la causa más habitual de muerte3.
La ELA produce una degeneración de la primera motoneurona en el córtex cerebral, que afecta con mayor gravedad a las neuronas de la circunvolución precentral que a las de la poscentral y que conlleva la degeneración walleriana del haz corticoespinal (HCE) y corticobulbar, desde la corteza motora, pasando por la cápsula interna, pedúnculos cerebrales, tronco del encéfalo y médula espinal1,3. Asimismo, también tiene lugar la degeneración de la segunda motoneurona en el asta anterior del cordón medular1,3. Secundariamente a la pérdida de neuronas motoras se produce una atrofia macro y microscópica tanto en las raíces nerviosas como en las circunvoluciones cerebrales afectadas, que se puede observar en estadios ya avanzados de la enfermedad y que por tanto no es útil a la hora de dar un diagnóstico de presunción cuando se presenta la sospecha clínica de ELA1,3.
Se trata por tanto de una degeneración de las neuronas de la corteza motora cerebral, de los núcleos motores tronco-encefálicos y del asta anterior de la médula, que condiciona la clínica, pudiendo presentar signos de paresia espástica, hiperreflexia y Babinski propios de las motoneuronas corticales, disartria y disfagia, propios de la parálisis bulbar, o debilidad, fasciculaciones y atrofia muscular progresiva, típicos de la lesión de las motoneuronas espinales o bien una combinación de todas ellas1,3,4. Otros signos y síntomas que podemos encontrar son dificultad para la marcha, pérdida de peso inexplicable, hipoxia e incluso arritmia cardíaca1. Característicamente no cursa con trastornos sensitivos, del control de esfínteres, ni afectación de la musculatura extrínseca ocular3.
El diagnóstico de ELA no presenta dificultad en fases evolucionadas de la enfermedad. En las fases iniciales, el diagnóstico se basa en la exploración clínica y la exclusión de otras enfermedades con síntomas parecidos3,5. Para la valoración de la motoneurona inferior nos servimos además de la electromiografía o del examen electrofisiológico, mientras que la valoración de la motoneurona superior constituye todavía un reto3,5.
Los criterios de El Escorial para el diagnóstico de ELA se sirven de la RM únicamente para descartar otras enfermedades con clínica similar que pudieran llevar a un diagnóstico de presunción erróneo5. Esto es así debido a que las técnicas convencionales de RM no permiten la demostración de la degeneración del HCE, a pesar de que ésta esté presente en los estudios de necropsia3. Aun así, la RM convencional tiene un papel fundamental en el diagnóstico inicial de la ELA, ya que puede reducir de forma importante el diagnóstico diferencial de los casos clínicamente sospechosos. En los casos de inicio bulbar o seudobulbar clínicamente claros, estos estudios de imagen no estarían siquiera indicados5.
Los diagnósticos que más frecuentemente simulan ELA (posible o probable) incluyen lesiones cerebrales (esclerosis múltiple y enfermedad cerebrovascular), lesiones de la base de cráneo, mielopatía cervical espondiloartrósica, otras mielopatías (lesiones del foramen magnum, tumores medulares y siringomielia) lesiones del cono medular, radiculopatía dorso-lumbo-sacra y enfermedades hereditarias como la enfermedad de Kennedy (atrofia muscular bulboespinal)3,5,6.
Existe un interés creciente sobre las técnicas de neuroimagen avanzadas de RM como la transferencia de magnetización (TM), la espectroscopia por RM (ERM), el tensor de difusión (TD) o la RM funcional (RMf) que recientemente han sido el foco de estudio en varias publicaciones y revisiones, todas ellas encaminadas a evaluar la fiabilidad diagnóstica de las mismas. Estas técnicas permiten identificar signos indirectos de neurodegeneración del HCE in vivo, aunque aún no existen suficientes evidencias que demuestren su utilidad en el diagnóstico inicial de la enfermedad. Según dichos estudios la ERM junto al TD parecen ser los métodos más prometedores para corroborar el diagnóstico clínico, con una alta especificidad y sensibilidad incluso en estadios tempranos de la enfermedad4,7,8.
Por otro lado las últimas investigaciones apuntan que la ELA no es únicamente un trastorno motor, sino un trastorno multisistémico, que varía tanto en sus formas de presentación como en las manifestaciones extramotoras que puede presentar9. Aunque los pacientes con ELA generalmente mantienen sus capacidades mentales hasta estadios terminales de la enfermedad, una pequeña proporción de ellos presentan además demencia (ELA-D)9,10. Estos pacientes presentan un tipo de demencia frontotemporal clínicamente diferente de la enfermedad de Alzheimer, cuyo origen se encuentra en la degeneración de los lóbulos frontal y temporal, y que se engloba dentro de las demencias frontotemporales10.
Se han encontrado ciertos hallazgos de imagen, que si bien no son específicos de ELA-D, pueden indicar este tipo de procesos neurodegenerativos. La atrofia cortical de los lóbulos temporales y frontal, unido a una línea de señal hiperintensa en T2 localizada en la sustancia blanca subcortical de la región anteromedial del lóbulo temporal, podrían ser útiles en el diagnóstico de la ELA-D9,10. Dicha hiperintensidad se correspondería histológicamente con una pérdida de mielina y axones asociada a gliosis10. La atrofia cortical se correspondería con cambios espongiformes, con pérdida neuronal y gliosis10.
Atrofia-volumetríaLos estudios volumétricos tanto globales como regionales también han sido utilizados en el estudio de los pacientes con ELA. Las técnicas de morfometría basada en vóxeles (MBV) permiten detectar precozmente la pérdida de volumen de la región del córtex motor primario y área premotora y están siendo incluidas en muchos ensayos clínicos. Estos estudios han demostrado atrofia de la sustancia gris más allá del córtex motor como los lóbulos parietales y región frontotemporal11,12. Esta atrofia es más marcada en los casos que se asocian a demencia frontotemporal y se correlaciona con los estudios histológicos que demuestran la existencia de inclusiones intraneuronales ubiquitina positivas características de este tipo de demencia13. También se ha demostrado la presencia de atrofia de la sustancia blanca de áreas extramotoras como el cuerpo calloso, el cerebelo y las áreas frontotemporales y occipitales14,15. Las medidas de área de la médula cervical parecen ser útiles para monitorizar la evolución de estos pacientes16.
No obstante las medidas cuantitativas de la atrofia cerebral se encuentran en un estadio preliminar de desarrollo, estandarización y normalización, lo que implica una importante dispersión de hallazgos y por tanto no las hace indicadas en la práctica clínica8,17.
Secuencia de transferencia de magnetizaciónEl contraste en la secuencia de TM se debe a la magnetización transversal de protones de agua unidos a macromoléculas rígidas (como la mielina) y la posibilidad de su transferencia a los protones móviles de agua libre tras un pulso de saturación. Las lesiones en las que se produce desmielinización, como ocurre en la ELA o en la esclerosis múltiple, provocan un significativo descenso de la transferencia de magnetización, dando lugar a una señal hiperintensa en las secuencias spin-echo (SE) potenciadas en T1 con TM. Esto también ocurre, aunque de manera menos marcada en las enfermedades que cursan con edema tisular18. De esta forma, esta técnica puede indicar la presencia de cambios estructurales en el tejido, que se asocian a distintas enfermedades, incluso cuando no son visibles con otras secuencias, y permite la realización de estudios cualitativos y/o cuantitativos4,18.
En el estudio de Rocha et al utilizando imágenes de TM potenciadas en SE T1 en los pacientes afectados por ELA se encontró hiperintensidad de señal a lo largo del HCE, que no se visualizaba en los sujetos sanos. La sensibilidad para la técnica de TM fue de un 80% y la especificidad del 100%. Estos autores atribuyen dichos hallazgos a la pérdida de mielina en los axones tras la degeneración somática cortical. No se observaron diferencias significativas, en cuanto a la imagen radiológica, entre los 2 hemisferios cerebrales, a pesar de que la clínica fuera más acusada en un hemicuerpo4.
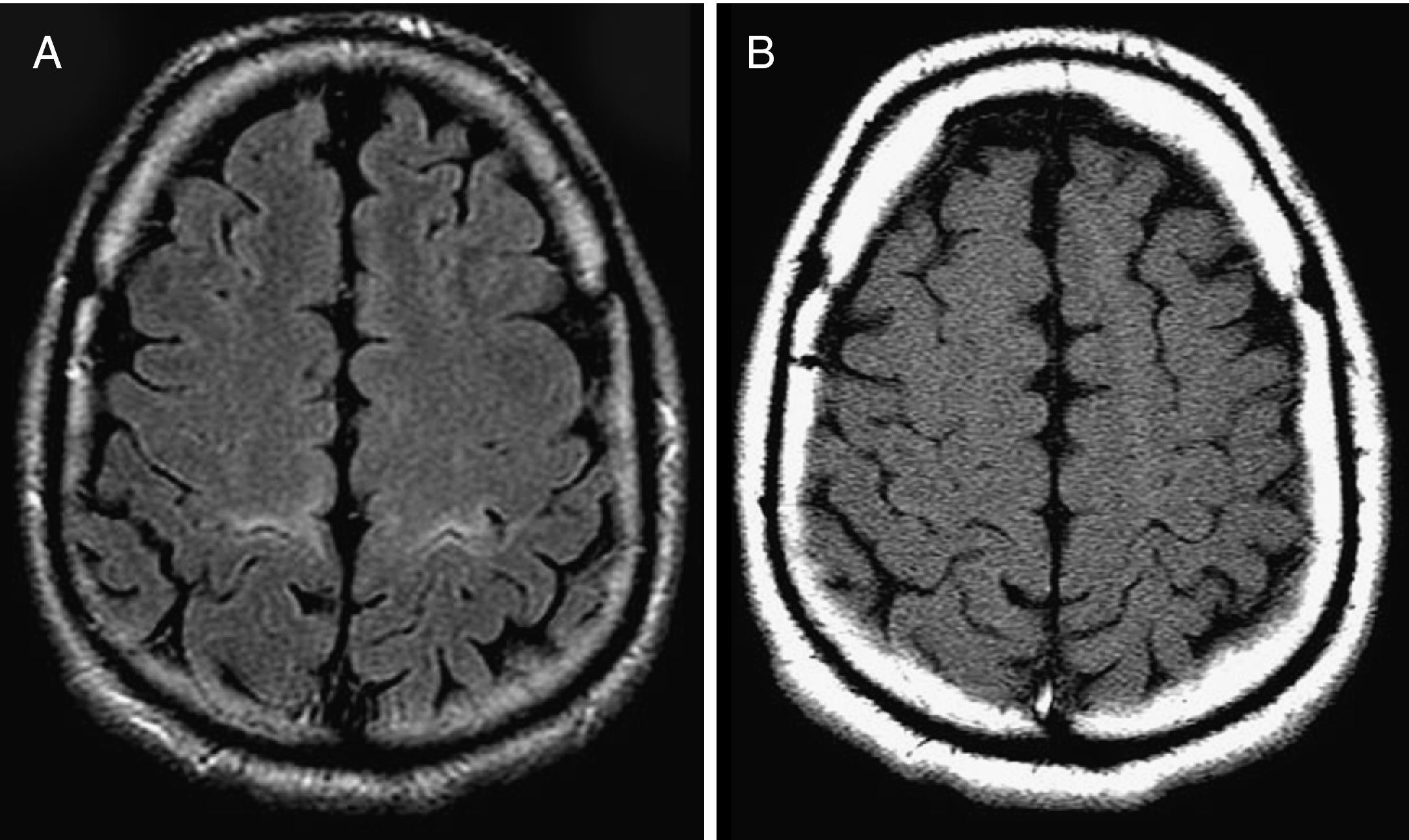
Diferentes grupos de investigadores han defendido que este hallazgo también se observa en las secuencias potenciadas en T2, densidad protónica (DP) y FLAIR (fluid attenuation inversion recovery)19–21, pero con el tiempo se ha demostrado que dicha señal hiperintensa en estas secuencias de RM convencional aparece con frecuencia en sujetos sanos y por tanto carece de especificidad4,21–23 (figs. 1 y 2).
. A) Se observó hiperintensidad de señal en la circunvolución precentral y en el resto del HCE bilateralmente (no mostrado), mientras que en las imágenes SE-T1 con pulso de TM la señal fue hipointensa (B) y por tanto no concluyente para ELA, como se demostró en la evolución clínica del paciente.")
Falso positivo en la secuencia FLAIR. Paciente de 53 años con clínica indicativa de ELA (debilidad de miembros inferiores de predominio derecho y fasciculaciones difusas). A) Se observó hiperintensidad de señal en la circunvolución precentral y en el resto del HCE bilateralmente (no mostrado), mientras que en las imágenes SE-T1 con pulso de TM la señal fue hipointensa (B) y por tanto no concluyente para ELA, como se demostró en la evolución clínica del paciente.
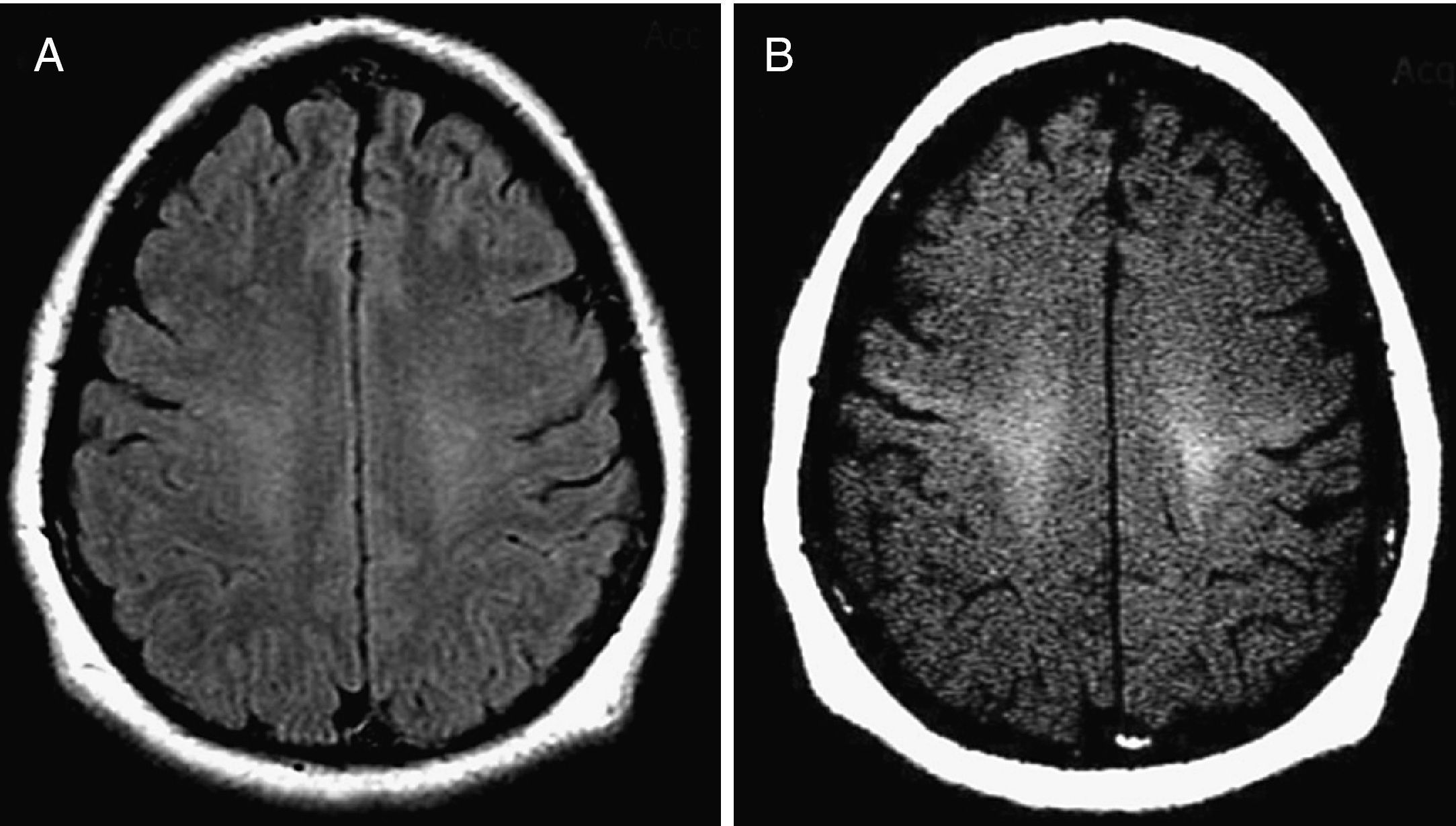
 y demencia frontotemporal (presente en la madre y 3 tíos maternos) que ingresa por trastornos de la marcha y del habla. A la exploración destaca un marcado tetrapiramidalismo, así como disartria y disfagia discretas. Además existían signos de denervación activa en miembros inferiores en el estudio neurofisiológico. Tanto la secuencia FLAIR (A), como la imagen SE potenciada en T1 con pulso de TM (B) mostraron una señal hiperintensa bilateral en el HCE desde el tronco del encéfalo hasta el área precentral correspondiente a la sustancia blanca bajo la corteza motora primaria y secundaria.")
Paciente con ELA clínicamente confirmada. Varón de 38 años con antecedentes familiares de ELA (2 tíos maternos fallecidos por dicha enfermedad) y demencia frontotemporal (presente en la madre y 3 tíos maternos) que ingresa por trastornos de la marcha y del habla. A la exploración destaca un marcado tetrapiramidalismo, así como disartria y disfagia discretas. Además existían signos de denervación activa en miembros inferiores en el estudio neurofisiológico. Tanto la secuencia FLAIR (A), como la imagen SE potenciada en T1 con pulso de TM (B) mostraron una señal hiperintensa bilateral en el HCE desde el tronco del encéfalo hasta el área precentral correspondiente a la sustancia blanca bajo la corteza motora primaria y secundaria.
Igualmente se ha descrito una señal hipointensa en la sustancia gris de la circunvolución precentral en las secuencias T2, T2* y FLAIR conocida como motor dark line en los pacientes con ELA4,19–22 (fig. 3A). Sin embargo, las últimas publicaciones demuestran que este patrón tampoco es específico de esta entidad, pudiéndose visualizar en personas sanas y también en pacientes con otras enfermedades neurodegenerativas4,19–22.
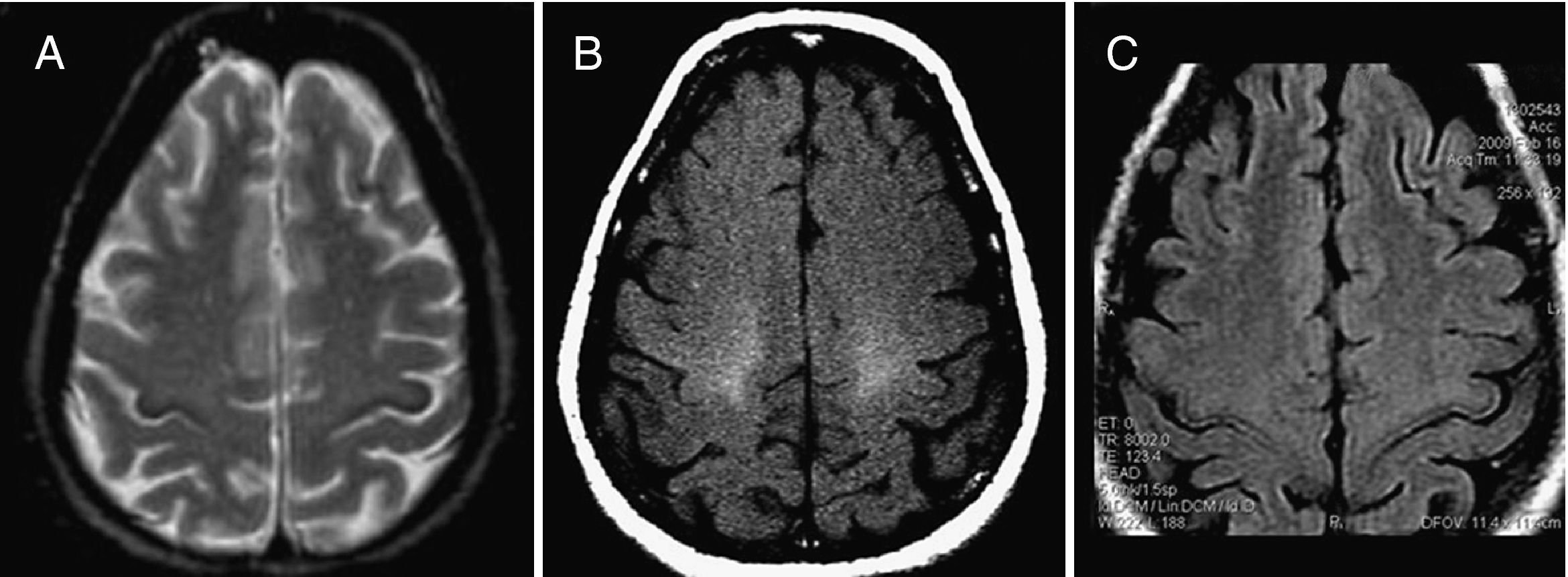
 En la secuencia potenciada en T2 se observa una hipointensidad giriforme a lo largo de la corteza de la circunvolución precentral (motor dark line). B) Exclusivamente en la secuencia TM existía un llamativo aumento de la señal del HCE (mostramos la región de los centros semiovales). C) En la secuencia FLAIR únicamente se pone de manifiesto un doble contorno hipointenso a lo largo de ambas cortezas motoras primarias, hallazgo característico pero menos específico.")
ELA confirmada. Paciente de 62 años que refiere debilidad progresiva y torpeza de movimiento que le impide realizar las actividades de la vida diaria. Posteriormente comienza con disfagia y disfonía. Destacaba la presencia de fasciculaciones en las 4 extremidades, reflejos plantares extensores muy vivos y una marcada denervación en el territorio de la motoneurona espinal y bulbar en el estudio neurofisiológico. A) En la secuencia potenciada en T2 se observa una hipointensidad giriforme a lo largo de la corteza de la circunvolución precentral (motor dark line). B) Exclusivamente en la secuencia TM existía un llamativo aumento de la señal del HCE (mostramos la región de los centros semiovales). C) En la secuencia FLAIR únicamente se pone de manifiesto un doble contorno hipointenso a lo largo de ambas cortezas motoras primarias, hallazgo característico pero menos específico.
En el estudio de Rocha et al el 24% de los pacientes con ELA mostraban esta motor dark line, pero también la presentaban el 19% del grupo control4. Asimismo Ngai et al han concluido que existe una relación significativa entre el envejecimiento y el aumento de frecuencia tanto de la hiperintensidad de la sustancia blanca subcortical precentral, como de la hipointensidad del córtex precentral en las secuencias FLAIR, confirmando la falta de especificidad de estos signos22.
En nuestra experiencia la TM es una secuencia útil para corroborar el diagnóstico clínico en los casos en que es positiva (fig. 3). En la actualidad no existen nuevas publicaciones de estudios cualitativos con TM que confirmen los hallazgos de Rocha et al. Sí se han realizado estudios cuantitativos, mediante la medida del ratio o cociente de magnetización transversal (MTR). La reducción en los valores de MTR se ha asociado con degeneración axonal y pérdida de mielina tanto en enfermedades inflamatorias del sistema nervioso central y esclerosis múltiple como en la enfermedad de motoneurona24,25. Los estudios de MTR aplicados a la ELA han sido discrepantes. En 2 de estos estudios se encontró una disminución del MTR en el HCE, que oscilaba entre el 2,6 y el 20% en los pacientes con ELA frente al grupo control, pero en la publicación más reciente de Charil et al no se encontraron diferencias significativas entre los casos y los controles. Se cree que en este último caso la gliosis reactiva a la pérdida axonal habría podido conducir en algunos casos a una seudonormalización del MTR12,25,26.
Espectroscopia por RMLa ERM es un método capaz de detectar y cuantificar los cambios neurodegenerativos tisulares mediante el análisis de los niveles de ciertos metabolitos presentes en el parénquima cerebral, que incluso podrían servir como marcadores de la progresión de la enfermedad o de la respuesta al tratamiento. Los metabolitos cuantificados han sido principalmente el N-acetil aspartato (NAA) marcador de la integridad neuronal, la creatina y fosfocreatina (Cr), marcadores de la capacidad energética y la colina (Cho), asociada a la síntesis de las membranas celulares27.
Pohl et al encontraron un descenso significativo de las concentraciones absolutas de NAA y de Cr, mientras que la Cho no sufría variaciones significativas28. Los cocientes NAA/Cho y NAA/Cr también estaban disminuidos y el Cho/Cr estaba elevado. La cuantificación del cociente NAA/Cho resultó ser el parámetro más útil a la hora de valorar la integridad de las motoneuronas en los pacientes con ELA, tanto en el diagnóstico como en el seguimiento, ya que fue el valor que más se modificó, presentando los mejores valores de especificidad y sensibilidad28–30. Este cociente presentaba un descenso significativo en el córtex contralateral al hemicuerpo con mayor afectación clínica y también una correlación débil aunque significativa entre gravedad y duración de la enfermedad. En cuanto al seguimiento se obtuvieron correlaciones significativas entre el empeoramiento de la sintomatología y el descenso de NAA/Cho y el aumento de Cho/Cr28.
Otros estudios como el de Mitsumoto et al también encontraron una marcada reducción de la concentración de NAA en la corteza motora primaria en los pacientes con ELA en relación con el grupo de controles, con el consecuente descenso del cociente NAA/Cr31.
Kaufmann et al observaron una asociación significativa entre el descenso del cociente NAA/Cr y la afectación de motoneuronas superiores. Concluyeron que la ERM mostraba una sensibilidad del 86% para detectar afectación de la primera motoneurona y una buena correlación con los signos clínicos, que unidos a otras técnicas diagnósticas, ayudarían al diagnóstico temprano de la ELA32.
Pohl et al encontraron además una aparente estabilización del cociente NAA/Cho en las fases en las que los pacientes no empeoraban clínicamente, pero al progresar la sintomatología este cociente sí variaba, sufriendo un descenso. Este hallazgo abriría la posibilidad de utilizar dicho parámetro como un marcador de actividad de la enfermedad28. La monitorización de los niveles de NAA en la corteza motora primaria y el HCE podría servir además como indicador de la respuesta al tratamiento8.
Según Pyra et al la circunvolución precentral seguida por la corona radiata constituyen las localizaciones en las que los cocientes NAA/Cho y NAA/Cr se encuentran más descendidos. Además la disminución de NAA/Cho en dicha circunvolución se correlacionaría con una progresión más rápida de la enfermedad33. La edad elevada, la duración breve de los síntomas y los niveles bajos de NAA/Cho se han relacionado con un mal pronóstico34.
El mecanismo de descenso de la Cr en la corteza cerebral de los pacientes con ELA todavía no está claro. Ha sido observado también en otras enfermedades neurodegenerativas como la enfermedad de Alzheimer, por lo que se ha especulado que su reducción podría indicar una alteración en el metabolismo celular y en el transporte de membrana de las células neuronales35,36. Esto apoyaría la teoría de que el daño oxidativo y la depleción de la adenosina trifosfato (ATP) pueden tener un papel importante en la patogenia de la ELA, ya que estos pacientes presentan alteraciones mitocondriales en las motoneuronas y en el músculo periférico35,36. Vielhaber et al llegaron a la conclusión de que la deficiencia de enzimas de la cadena respiratoria mitocondrial afectaba a la supervivencia de las motoneuronas corticales. Esto es lo que ocurriría en los sujetos afectados por ELA, donde el NAA, metabolito producido en la mitocondria, se encuentra disminuido36.
Por otro lado, el daño neuronal se acompaña de un aumento de la actividad glial, que en el estudio espectroscópico se refleja en un aumento del mioinositol (mIns)8,37,38. Este hallazgo ha sido demostrado también en la corteza motora de los pacientes con ELA, donde el cociente NAA/mIns se encuentra disminuido34,37,39. Este cociente parece ser el más sensible (75%) y específico (93%) en la detección de la ELA en los estudios de ERM que utilizaron equipos con campo magnético de 3 teslas, siendo también el parámetro que más se correlacionó con la gravedad de la enfermedad34,37.
En conclusión, según la bibliografía, en la valoración de la afectación de la primera motoneurona es de mayor utilidad el estudio de los cocientes de metabolitos que la cuantificación de los mismos individualmente, siendo característico el descenso de los cocientes NAA/mIns, NAA/Cho, NAA/Cr y NAA/Cho+Cr en la corteza motora. El cociente NAA/mIns sería el más útil en la detección de la enfermedad ya que según los estudios de Kalra et al es el que más modificaciones sufre34,37, mientras que el seguimiento del descenso de NAA en la corteza motora primaria y en el HCE podría ser de utilidad en la evaluación de la progresión y de la respuesta al tratamiento8.
Tensor de difusiónOtra técnica de RM que está siendo utilizada para proporcionar información cuantitativa acerca de la pérdida axonal que se produce en la vía piramidal en los pacientes afectados de ELA es el tensor de difusión (TD)29,40–44. El TD permite la visualización y evaluación inicial del HCE, siempre que se realice de forma grupal y no individual, proporcionando una información indirecta del daño de la motoneurona superior41,42,44. Esta técnica se basa en que en las fibras de sustancia blanca, al contrario de lo que ocurre en la sustancia gris, las moléculas de agua no se mueven con la misma libertad en todas las direcciones, sino que su movilidad está menos restringida en sentido paralelo a las fibras. A este fenómeno se le conoce como difusión anisotrópica. Por lo tanto, las alteraciones que se producen en la difusión de las moléculas de agua de los tejidos, como las que ocurren en la degeneración del HCE, son detectadas por el TD, mostrándonos en imagen los cambios resultantes en la citoarquitectura de esta vía41.
Mediante este método se obtienen mapas en color que pueden ser estudiados en distintos planos de corte haciendo posible un análisis volumétrico, constituyendo por tanto un potencial método de monitorización de los cambios morfológicos del HCE (mapas de anisotropía fraccional con codificación de color)40.
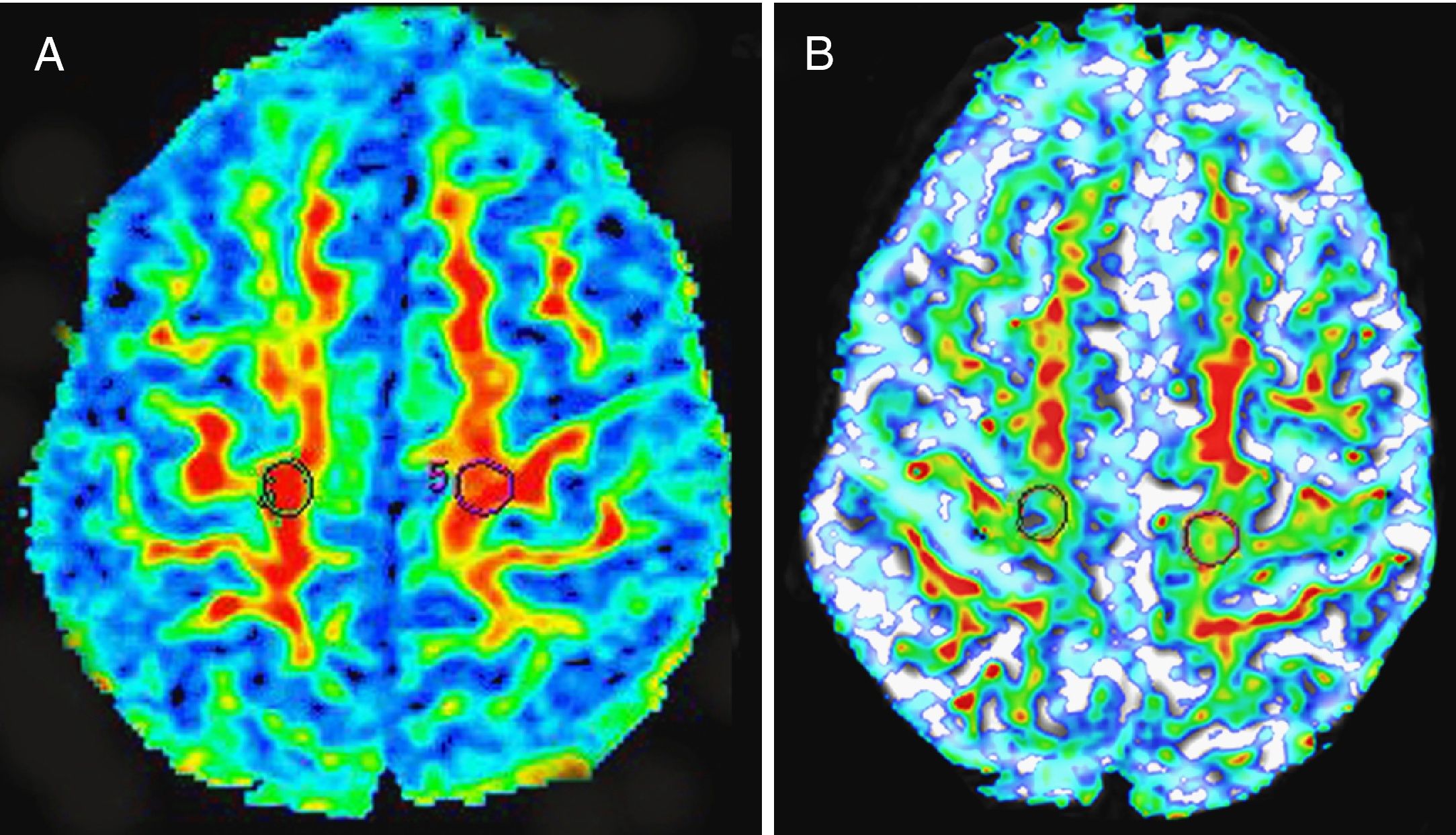
El parámetro más utilizado es la fracción de anisotropía (FA), con el que podemos medir la variación en la anisotropía del tejido en las diferentes enfermedades. En la ELA, parece existir una disminución de la FA en el HCE, que representa la pérdida de integridad de sus fibras producidas por la neurodegeneración característica de esta enfermedad42–52.
Se ha observado además una correlación entre esta reducción de la FA y una mayor afectación clínica42,44–47, una progresión más rápida de la enfermedad52 y los datos electrofisiológicos de afectación de la motoneurona superior43. Además los pacientes en los que la enfermedad se inicia con afectación bulbar parecen tener las cifras de FA más alteradas46.
La FA también parece estar significativamente disminuida en la médula cervical de los pacientes con ELA, presentando una fuerte correlación con la gravedad de la enfermedad53.
La difusibilidad media (DM) es otro parámetro que podemos medir en las secuencias de TD, la cual se ve aumentada en el HCE de los pacientes con ELA45–52 y que parece correlacionarse con la duración de la enfermedad45,47.
En varios de los estudios realizados el brazo posterior de la cápsula interna fue la localización donde se encontraron los valores más alterados de FA y DM45–52.
Otra técnica de imagen derivada del TD es la tractografía. Se trata de una técnica de posprocesado (con diferentes programas de software disponibles) que muestra la localización y trayectoria de los haces axonales mediante un algoritmo matemático y un método probabilístico40. El análisis volumétrico mediante tractografía no ha podido ser validado aún, ya que es dependiente del usuario, además de verse influido por otras muchas variables29,40. Aun así parece ser que el volumen del HCE en los pacientes con ELA está disminuido comparativamente con la población sana.
Wang et al40 realizaron un análisis volumétrico del HCE sobre mapas de colores, en 16 casos y 17 controles, encontrando que el volumen de este haz era significativamente menor en pacientes con ELA en relación con la población sana. No encontraron diferencias significativas en el volumen del HCE en el hemisferio con mayor afectación en relación al menos afectado.
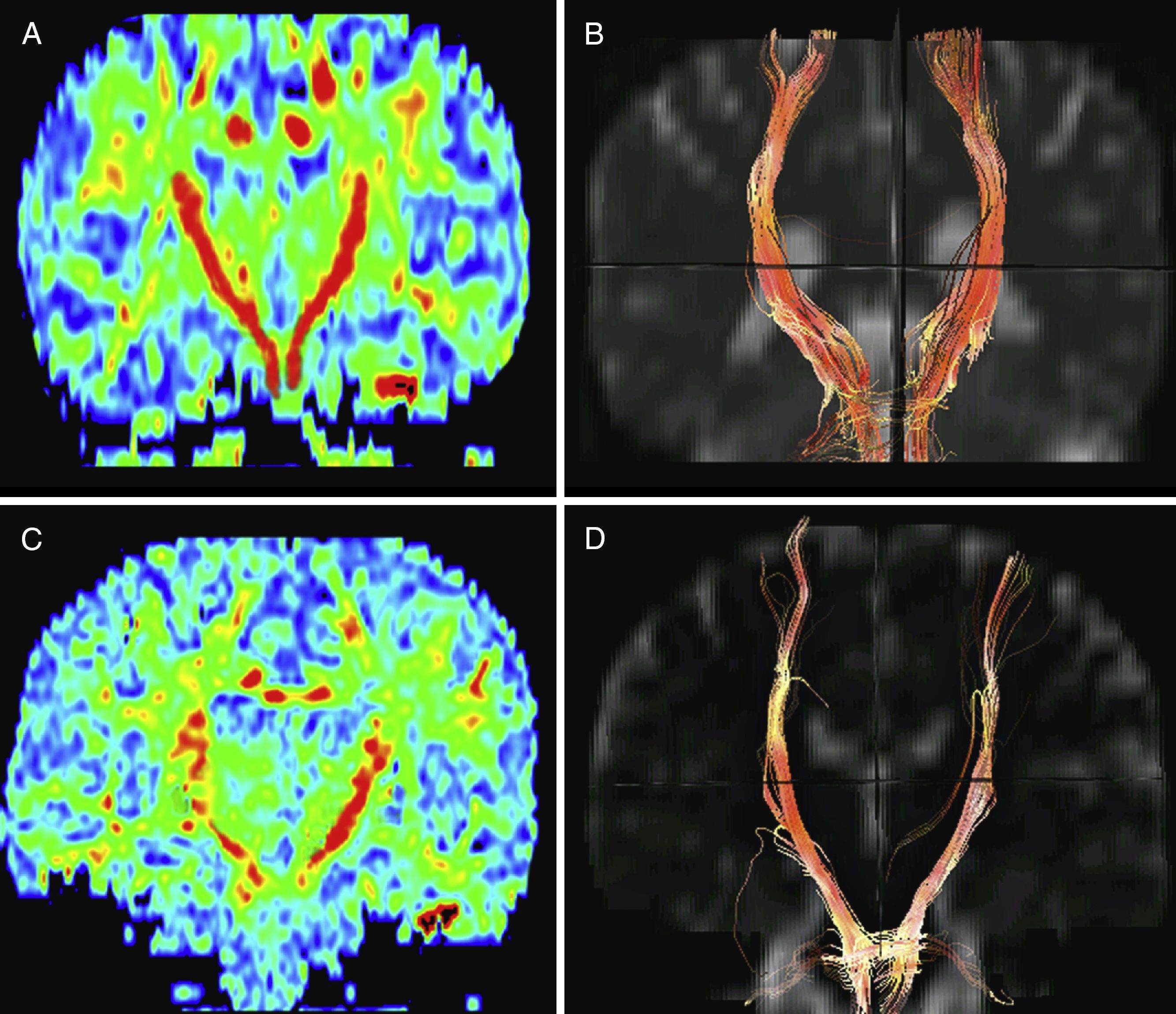
Por esta razón aunque el TD puede detectar daños cerebrales incluso más precozmente que otras pruebas de imagen, debido a su difícil validación y al intenso trabajo de posprocesado que requiere, no ha sido incluido por el momento en la mayoría de los protocolos de diagnóstico ante la sospecha de ELA29,38. Dicha técnica sí podría ser utilizada en el seguimiento de la enfermedad y en la valoración de la respuesta individual al tratamiento, aunque su uso se encuentra todavía limitado a ensayos clínicos44,52 (figs. 4–6).
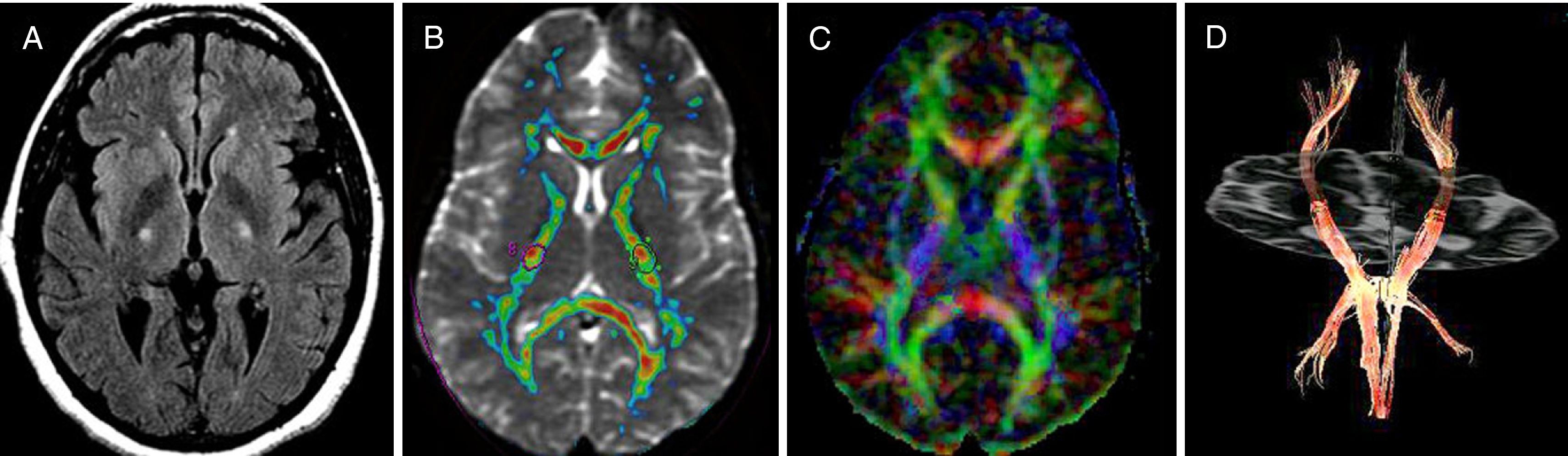
. La anisotropía fraccional y la difusibilidad media medidas mediante la colocación de ROI estaba disminuida y aumentada respectivamente en el brazo posterior de la cápsula interna (B), que se representa en color azul en la imagen estructural de TD debido a su dirección craneo-caudal predominante (C). La tractografía mostró una asimetría de los haces cortico-espinales con un aparente menor número de fibras en el lado izquierdo más afectado (D).")
ELA confirmada. Paciente de 65 años con debilidad progresiva y torpeza de movimientos. También presentaba disfagia y disfonía leve. En la exploración existía espasticidad y reflejo de Babinski positivo. En las imágenes de RM se observó una hiperintensidad a lo largo del HCE en la secuencia FLAIR bien patente a nivel de la cápsula interna (A). La anisotropía fraccional y la difusibilidad media medidas mediante la colocación de ROI estaba disminuida y aumentada respectivamente en el brazo posterior de la cápsula interna (B), que se representa en color azul en la imagen estructural de TD debido a su dirección craneo-caudal predominante (C). La tractografía mostró una asimetría de los haces cortico-espinales con un aparente menor número de fibras en el lado izquierdo más afectado (D).
 de volumen y FA normales (A). La tractografía del mismo paciente sano es normal (B). El mapa de color de TD en el paciente con ELA muestra una clara disminución de la anisotropía fraccional de ambos HCE más marcada en el lado derecho (C), así como también de su volumen, como muestra la tractografía, donde se observan unos haces corticoespinales más adelgazados con respecto al control normal (D).")
Sujeto sano de control y paciente con ELA de 63 años. En los mapas de color de TD reconstruidos en coronal se muestra el HCE en rojo (dirección cráneo-caudal) de volumen y FA normales (A). La tractografía del mismo paciente sano es normal (B). El mapa de color de TD en el paciente con ELA muestra una clara disminución de la anisotropía fraccional de ambos HCE más marcada en el lado derecho (C), así como también de su volumen, como muestra la tractografía, donde se observan unos haces corticoespinales más adelgazados con respecto al control normal (D).
. El mapa de color de TD del paciente con ELA muestra una clara disminución de la anisotropía fraccional de ambos HCE (B).")
Mapas de color de anisotropía fraccional en axial de un sujeto sano y un paciente con ELA. En el sujeto sano se observa una anisotropía de los haces corticoespinales normales señalados con ambos ROI (A). El mapa de color de TD del paciente con ELA muestra una clara disminución de la anisotropía fraccional de ambos HCE (B).
En los pacientes con ELA se han demostrado cambios adaptativos secundarios a la pérdida neural y desmielinización del HCE, que han sido explicados sobre la base de fenómenos de plasticidad y reorganización neuronal. Los estudios de RM funcional (RMf) en estos pacientes permiten estimar dichos cambios mediante la técnica BOLD (blood oxigenation level dependency), detectando aumentos significativos de la actividad neuronal en la corteza somatosensorial, corteza parietal inferior, corteza motora primaria tanto contralateral como ipsolateral al movimiento en tareas motoras y también en la corteza motora y premotora suplementarias bilateralmente54. Estos aumentos de la actividad evolucionan en el tiempo y parecen estar relacionados con tareas de programación, ejecución y preparación del movimiento. Este hecho se explica probablemente por fenómenos de desinhibición de conexiones transhemisféricas y reclutamiento de áreas premotoras suplementarias adicionales cuando progresa la enfermedad55. Además, se ha demostrado una mayor actividad en los ganglios basales, cerebelo, hipocampos y las amígdalas. La disfunción de los patrones de activación normales también se hace patente con paradigmas de activación de las áreas del lenguaje. Así, se ha encontrado una reducción de la activación de las circunvoluciones frontales media e inferior, cíngulo anterior y lóbulos parietal y temporal durante tareas de fluencia verbal y de Broca y regiones parieto-temporales en tareas de denominación56. La dificultad de controlar y estandarizar las tareas de activación así como las técnicas de posprocesado hace que exista una gran variabilidad en los estudios de RMf por lo que será necesario una normalización en la adquisición y en el análisis estadístico de dichos estudios8,38.
ConclusionesLas secuencias convencionales de RM (T1, T2 o FLAIR) tienen una escasa sensibilidad y especificidad en la detección de la afectación de la primera motoneurona en la ELA, especialmente en las fases iniciales de la enfermedad. La combinación de técnicas avanzadas como la ERM, la TM, la RMf, las técnicas volumétricas y las imágenes de TD está demostrando ser cada vez más útil en la detección de los pacientes con ELA, incrementando significativamente la sensibilidad y especificidad de las técnicas convencionales. La secuencia SE-T1 con TM por su fácil adquisición y sencilla interpretación es útil en el protocolo inicial, incrementando la sensibilidad de las técnicas convencionales en muchos casos. El MTR pese a ser una técnica sensible y cuantificable es más compleja de obtener y los resultados son muy variables. La ERM puede ser una herramienta útil especialmente en el seguimiento de la enfermedad mediante la monitorización y cuantificación de los ratios NAA/Cr, NAA/Cho y NAA/mIns. Cuando se combina con los hallazgos del TD se mejora la fiabilidad diagnósica. El TD permite la visualización y evaluación del HCE de forma grupal y no de forma individual. El propósito futuro de esta técnica es lograr una estandarización de los métodos de valoración y cuantificación de los mapas de AF y de la tractografía para que su interpretación y análisis sean fácilmente reproducibles. Las técnicas de RMf permitirán hacer un seguimiento de los cambios funcionales que se producen en el curso de la enfermedad independientemente de los cambios morfológicos.
En los próximos años el principal objetivo de estas técnicas será intentar aportar la suficiente sensibilidad y especificidad diagnóstica que permita confirmar de forma precoz aquellos casos con sospecha clínica de ELA. Así mismo, dichas técnicas combinadas podrían ser muy útiles en el control evolutivo del daño neuronal y en la monitorización de las diferentes terapias que vayan surgiendo siendo integradas progresivamente en la práctica clínica diaria.
AutoríasM.J. Álvarez-Uría Tejero: concepción y diseño del estudio, obtención de datos o análisis e interpretación del mismo, 50%; redacción del trabajo o revisión crítica, haciendo aportaciones intelectuales relevantes, 40%.
A. Sáiz Ayala: concepción y diseño del estudio, obtención de datos o análisis e interpretación del mismo, 20%; redacción del trabajo o revisión crítica, haciendo aportaciones intelectuales relevantes, 30%.
C. Fernández Rey: concepción y diseño del estudio, obtención de datos o análisis e interpretación del mismo, 12%; redacción del trabajo o revisión crítica, haciendo aportaciones intelectuales relevantes, 12%.
E. Santamaría Liébana: concepción y diseño del estudio, obtención de datos o análisis e interpretación del mismo, 10%; redacción del trabajo o revisión crítica, haciendo aportaciones intelectuales relevantes, 10%.
S. Costilla García: concepción y diseño del estudio, obtención de datos o análisis e interpretación del mismo, 8%; redacción del trabajo o revisión crítica, haciendo aportaciones intelectuales relevantes, 8%.
Todos los autores han leído y aprueban la versión final del artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

