





Suplemento “Patología Intersticial Pulmonar”
Más datosLas enfermedades quísticas pulmonares engloban a un grupo heterogéneo de entidades, caracterizadas por la presencia de quistes, correspondientes a lesiones pulmonares redondeadas de contenido usualmente aéreo y de pared fina. Su diagnóstico diferencial constituye un reto, que debe de ser manejado desde una perspectiva clínica y también radiológica. Entidades como el enfisema pulmonar y las bronquiectasias quísticas pueden simular enfermedades quísticas.
La tomografía computarizada de alta resolución es el método de imagen de elección en la evaluación y el diagnóstico de las enfermedades quísticas pulmonares, ya que confirma la presencia de enfermedad pulmonar y establece el correcto diagnóstico de las complicaciones asociadas. En muchos casos, el diagnóstico se establecerá preferentemente con base en los hallazgos presentes en esta técnica de imagen, obviando la necesidad de una comprobación anatomopatológica. Por estas razones, el radiólogo debe familiarizarse con las diferentes presentaciones de estas entidades.
Una amplia variedad de enfermedades se caracteriza por la presencia de quistes pulmonares difusos. Entre ellas, las más frecuentes son la linfangioleiomiomatosis, asociada o no a esclerosis tuberosa, la histiocitosis de células de Langerhans y la neumonía intersticial linfoide. Otras entidades menos frecuentes son el síndrome de Birt-Hogg-Dubé, la amiloidosis y la enfermedad por depósito de cadenas ligeras.
En este artículo se describen las características y las formas de presentación de algunas de estas entidades, haciendo hincapié en los detalles diferenciales de las mismas.
The term cystic lung disease encompasses a heterogeneous group of entities characterized by round lung lesions that correspond to cysts with fine walls, which usually contain air. The differential diagnosis of these lesions can be challenging, requiring both clinical and radiological perspectives. Entities such as pulmonary emphysema and cystic bronchiectasis can simulate cystic disease.
High-resolution computed tomography (HRCT) is the imaging technique of choice for the evaluation and diagnosis of cystic lung disease, because it confirms the presence of lung disease and establishes the correct diagnosis of the associated complications. In many cases, the diagnosis can be established based on the HRCT findings, thus making histologic confirmation unnecessary. For these reasons, radiologists need to be familiar with the different presentations of these entities.
A wide variety of diseases are characterized by the presence of diffuse pulmonary cysts. Among these, the most common are lymphangioleiomyomatosis, which may or may not be associated with tuberous sclerosis, Langerhans cell histiocytosis, and lymphocytic interstitial pneumonia. Other, less common entities include Birt-Hogg-Dubé syndrome, amyloidosis, and light-chain deposit disease.
This article describes the characteristics and presentations of some of these entities, emphasizing the details that can help differentiate among them.
Las enfermedades quísticas pulmonares son infrecuentes y se caracterizan en la tomografía computarizada (TC) por la presencia de quistes, definidos como lesiones redondeadas de baja atenuación, con contenido aéreo (ocasionalmente líquido o sólido), con una pared fina (< 3mm) constituida por tejido epitelial o fibrosis, y bien delimitadas por pulmón normal adyacente1. Es importante distinguirlos de otras lesiones pulmonares mucho más frecuentes que también contienen aire2–4:
- 1.
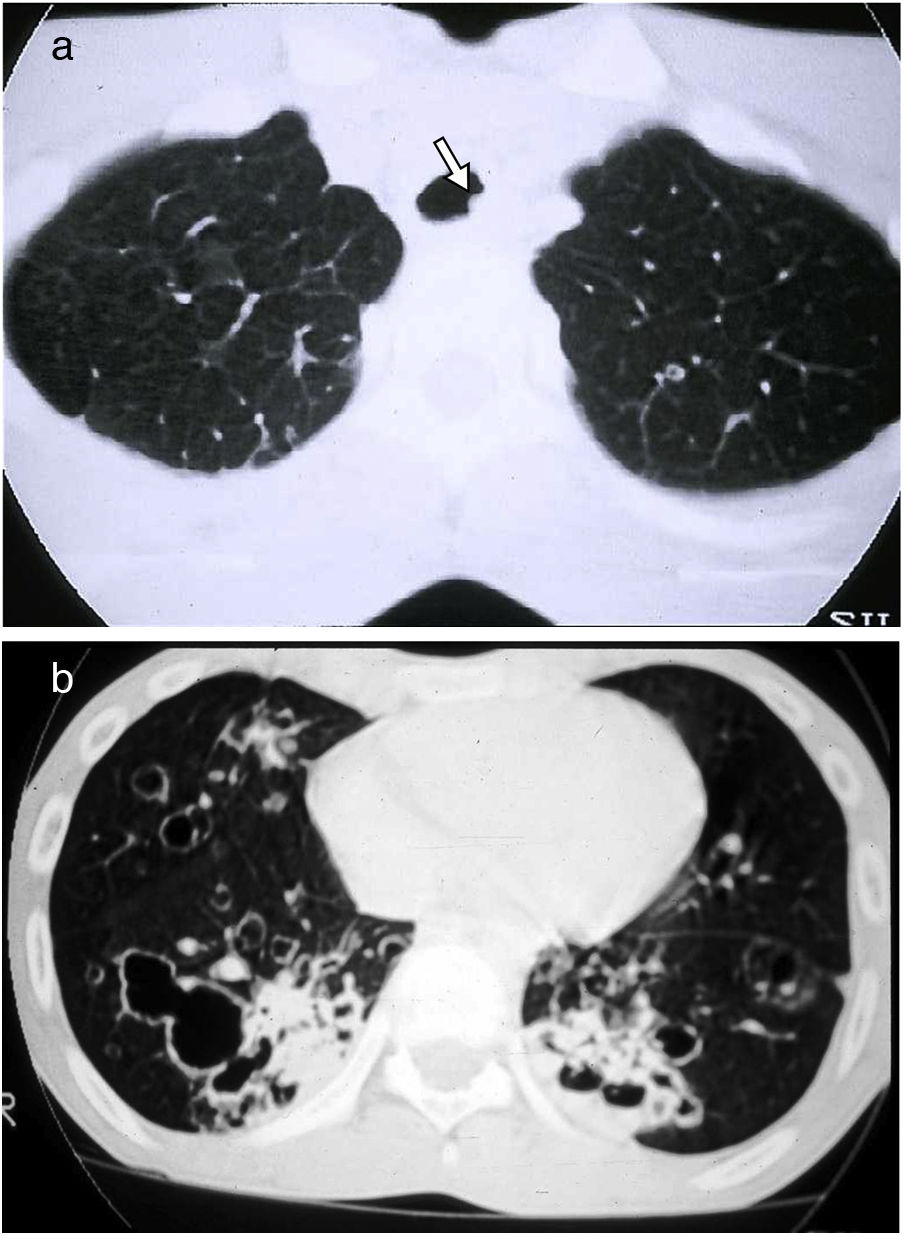
Cavidades: aparecen en el seno de consolidaciones, masas o nódulos y tienen una pared más gruesa (> 4mm) que la de los quistes (fig. 1).
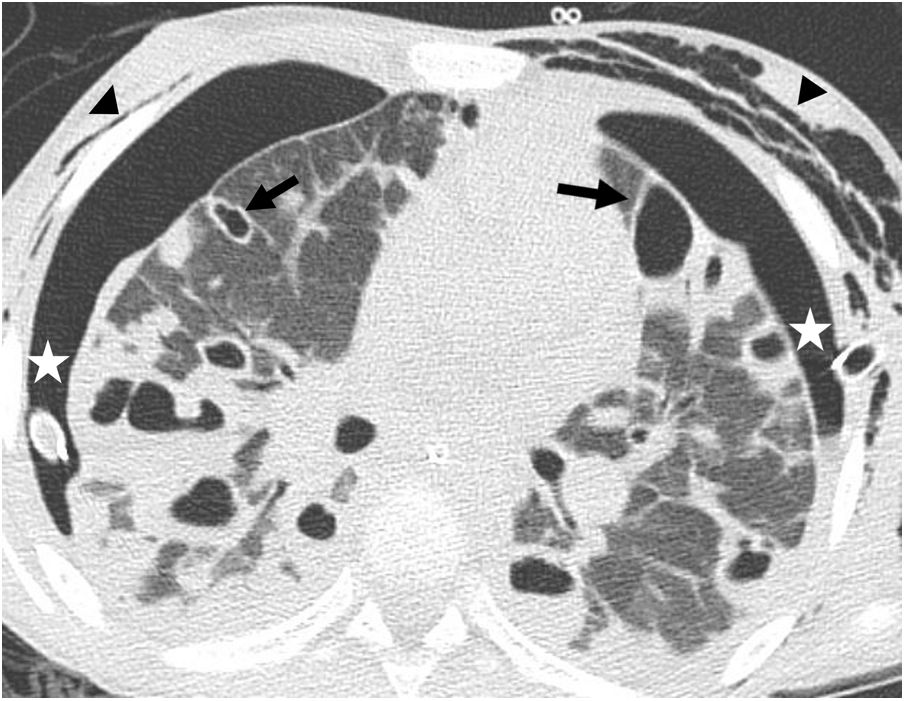
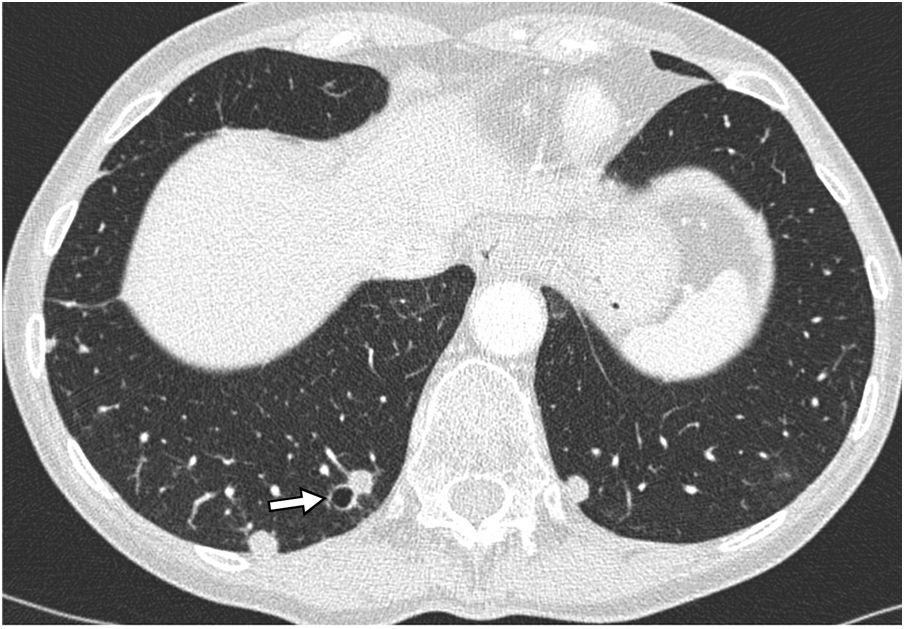
. Se observa neumotórax bilateral, presumiblemente debido a la rotura de alguna de las lesiones cavitadas periféricas, con tubos de drenaje pleural en ambos hemitórax (astericos). Se identifica además enfisema subcutáneo bilateral (puntas de flecha).") Figura 1.
Figura 1.Niña de 9 años con neumonía por Staphylococcus aureus. La imagen de TC muestra múltiples focos de cavitación aérea en el seno de nódulos pulmonares bilaterales, de distribución predominantemente subpleural, y en áreas de consolidación del lóbulo inferior derecho. Además, se ven quistes de pared fina bilaterales correspondientes a neumatoceles (flechas negras). Se observa neumotórax bilateral, presumiblemente debido a la rotura de alguna de las lesiones cavitadas periféricas, con tubos de drenaje pleural en ambos hemitórax (astericos). Se identifica además enfisema subcutáneo bilateral (puntas de flecha).
(0.11MB). - 2.
Enfisema centroacinar: áreas de disminución de la atenuación, sin pared, de hasta 1cm. En ocasiones se ve una pared fina y entran en el diagnóstico diferencial de las enfermedades quísticas pulmonares, pero la localización predominante en los campos superiores, la historia de tabaquismo y la frecuente identificación de un punto central que corresponde a la arteria centrolobulillar ayudan al diagnóstico (fig. 2).
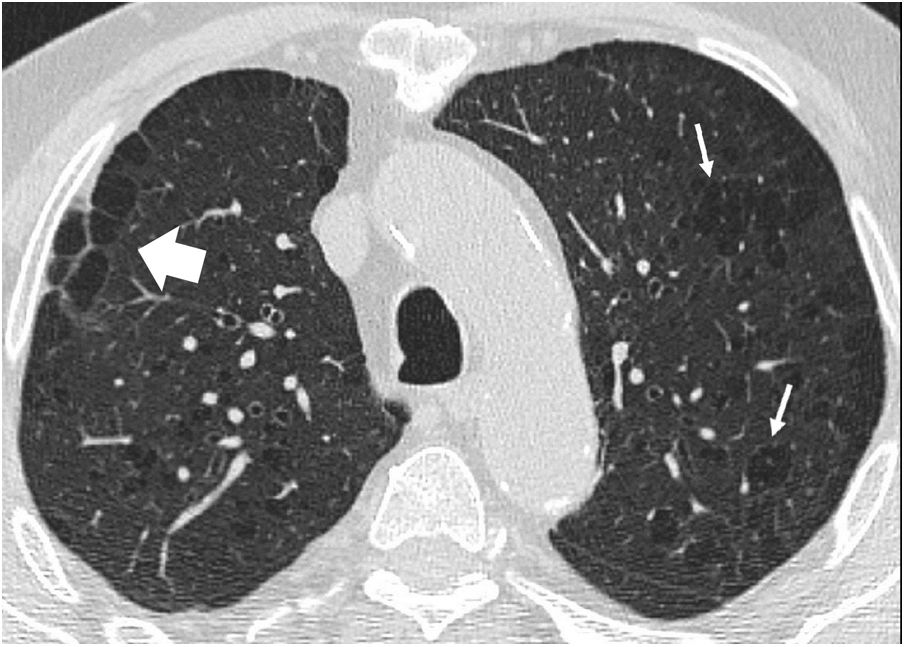
. Se aprecia también enfisema paraseptal con bullas subpleurales, sobre todo en el lóbulo superior derecho (flecha gruesa).") Figura 2.
Figura 2.Enfisema pulmonar centroacinar y paraseptal en un paciente fumador. La imagen de TCAR de los lóbulos superiores muestra múltiples áreas de disminución de la atenuación, sin pared apreciable, correspondientes a enfisema centroacinar. Destaca en algunas de ellas la presencia de la arteriola centrolobulillar en su interior (flechas). Se aprecia también enfisema paraseptal con bullas subpleurales, sobre todo en el lóbulo superior derecho (flecha gruesa).
(0.09MB). - 3.
Enfisema paraseptal y bullas: el enfisema paraseptal se manifiesta por áreas de baja atenuación subpleurales o peribroncovasculares de hasta 1cm de diámetro, limitadas por la pleura o septos, sin una verdadera pared como los quistes. Las bullas se deben a la confluencia de áreas de enfisema paraseptal y son lesiones>1cm, de predominio subpleural y con una pared casi imperceptible (fig. 2).
- 4.
Bronquiectasias quísticas: la revisión cuidadosa de los cortes axiales contiguos o de las reconstrucciones coronales o sagitales ayudan a reconocer su aspecto tubular. Pueden acompañarse de un engrosamiento de la pared bronquial, atrapamiento aéreo y nódulos con distribución de árbol en brote (fig. 3).
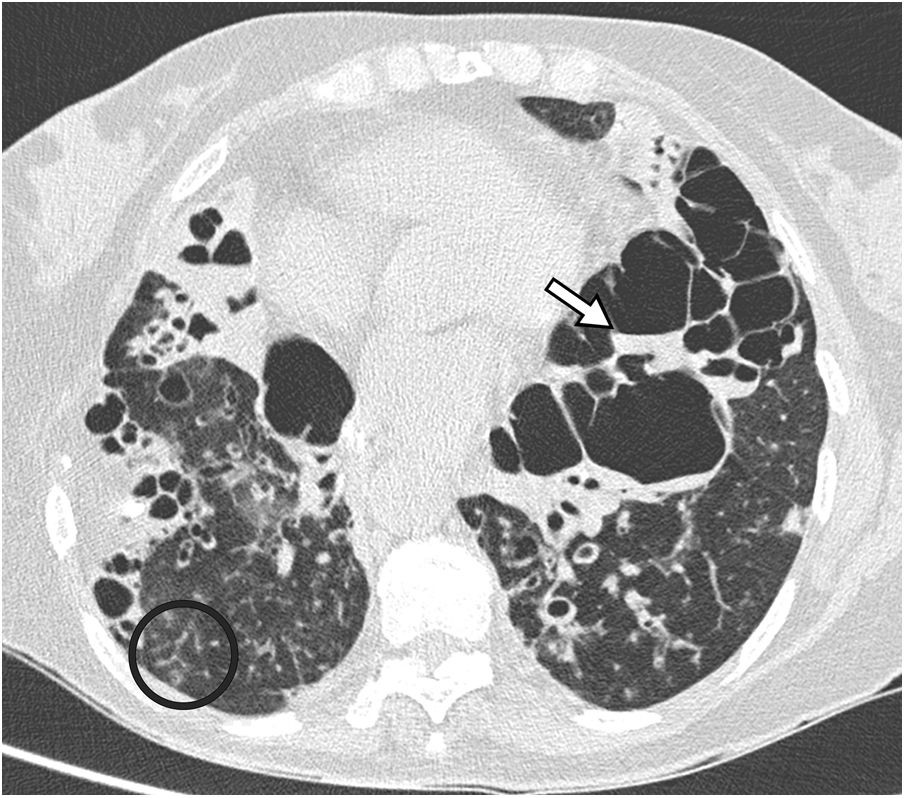
, correspondientes a bronquiectasias quísticas. Se observa también la presencia de nódulos con distribución de árbol en brote, correspondientes a impactaciones bronquiolares (círculo).") Figura 3.
Figura 3.Bronquiectasias quísticas sobreinfectadas. La imagen de TCAR de los lóbulos inferiores muestra múltiples imágenes quísticas bilaterales, algunas de ellas con un nivel hidroaéreo (flecha), correspondientes a bronquiectasias quísticas. Se observa también la presencia de nódulos con distribución de árbol en brote, correspondientes a impactaciones bronquiolares (círculo).
(0.14MB). - 5.
Neumatoceles: espacios aéreos redondeados de pared fina, secundarios a neumonía, traumatismo o aspiración de hidrocarburos, habitualmente escasos en número y transitorios, con resolución en semanas o meses (fig. 1).
- 6.
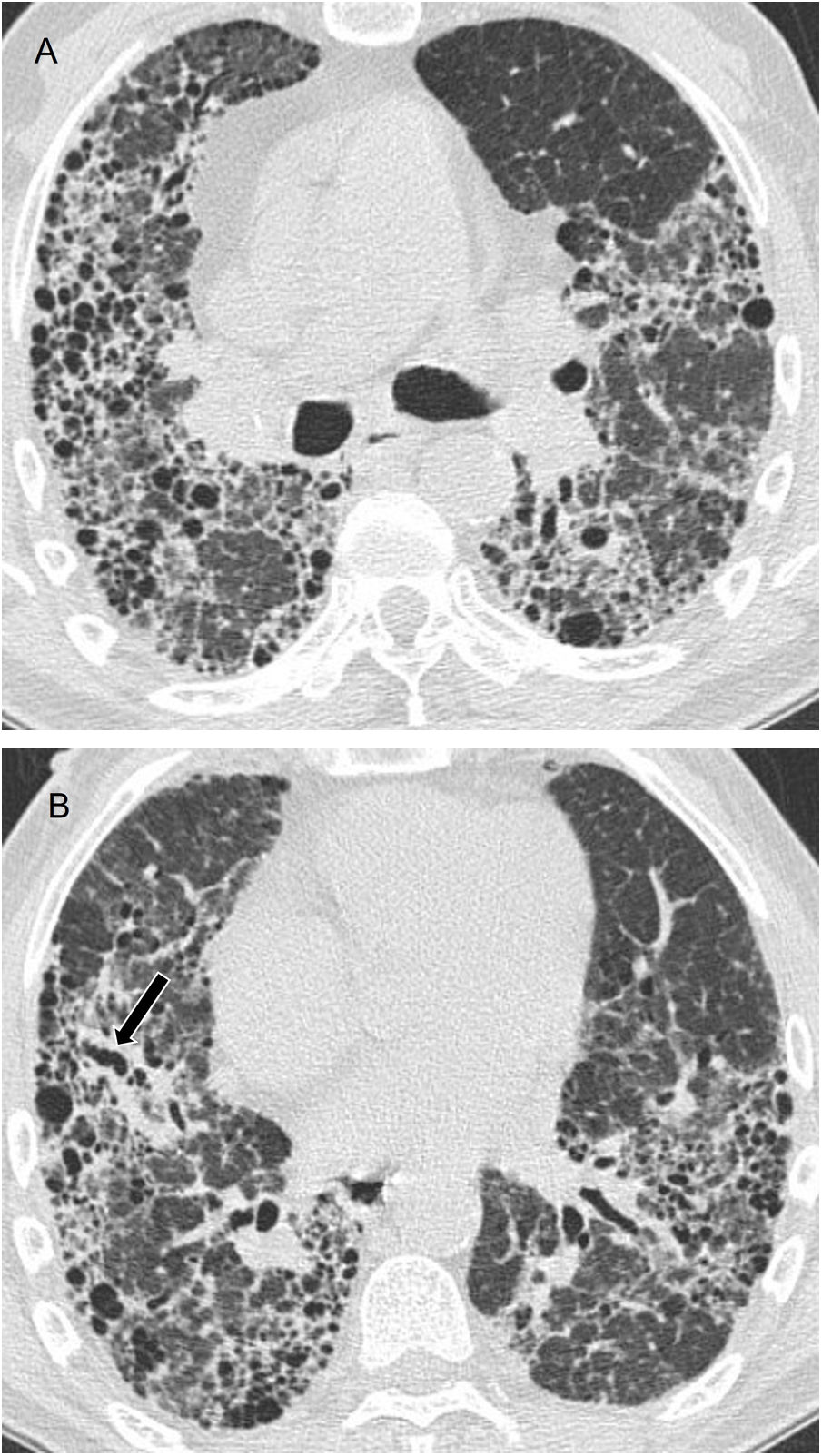
Los quistes de panal son lesiones quísticas aéreas con un tamaño habitualmente uniforme (3-10mm, ocasionalmente mayores), con una pared bien definida de 1-3mm, agrupadas de manera que comparten pared. En fases avanzadas se disponen en capas, típicamente en la región subpleural. Se consideran específicos de fibrosis pulmonar y se asocian a otros signos como pérdida de volumen, patrón reticular y bronquiectasias de tracción (fig. 4).
. Se observa neumotórax bilateral, presumiblemente debido a la rotura de alguna de las lesiones cavitadas periféricas, con tubos de drenaje pleural en ambos hemitórax (astericos). Se identifica además enfisema subcutáneo bilateral (puntas de flecha).")
. Se aprecia también enfisema paraseptal con bullas subpleurales, sobre todo en el lóbulo superior derecho (flecha gruesa).")
, correspondientes a bronquiectasias quísticas. Se observa también la presencia de nódulos con distribución de árbol en brote, correspondientes a impactaciones bronquiolares (círculo).")
 La imagen de TCAR muestra múltiples quistes subpleurales de pequeño tamaño, dispuestos en capas. B) En un corte más caudal se identifica una bronquiectasia de tracción (flecha).")
En la tabla 1 se recogen las principales enfermedades asociadas a quistes pulmonares. Para el diagnóstico diferencial de las enfermedades quísticas pulmonares es de especial importancia analizar el tamaño, el número, la morfología y la distribución de los quistes, junto con otros posibles hallazgos de imagen asociados, como opacidades en vidrio deslustrado o nódulos (tabla 2); en ocasiones, los hallazgos en la TC, junto con el contexto clínico, permitirán un diagnóstico específico, otras veces será necesaria la biopsia3–6.
Causas de enfermedades pulmonares quísticas difusas
| Causas principales |
| Linfangioleiomiomatosis |
| Histiocitosis pulmonar de células de Langerhans |
| Síndrome de Birt-Hogg-Dubé |
| Neumonía intersticial linfoide |
| Amiloidosis |
| Otras causas |
| Neumonitis por hipersensibilidad |
| Neumonía intersticial descamativa |
| Asociados al envejecimiento (Aging lung cysts) |
| Enfermedad por depósito de cadenas ligeras |
| Síndromes hereditarios: neurofibromatosis 1, síndrome de Proteus, síndrome de Ehler-Danlos |
| Malformación congénita de la vía aérea |
| Metástasis |
| Infecciones |
| Fibrosis avanzada (FPI, sarcoidosis, etc.) |
Diagnóstico diferencial de las principales enfermedades pulmonares quísticas difusas en la TC
| Enfermedad | Pared del quiste | Morfología de los quistes | Distribución | Hallazgos asociados |
|---|---|---|---|---|
| LLM | Fina | Redondeados | Difusa | Neumotórax, derrame pleural, linfangiomas, angiomiolipomas renales, ascitis |
| HCL | Fina/gruesa | Redondeados, irregulares o con forma «extraña» | Predominio en los campos superiores, respetan los ángulos costofrénicos | Nódulos pulmonares, algunos cavitados |
| BHD | Fina | Redondos, ovalados, lenticulares, septados | Predominio basal; distribución subpleural, paramediastínica, yuxtacisural | Neumotórax. Tumores renales |
| NIL | Fina | Redondeados | Difusos o en campos inferiores; peribroncovasculares, subpleurales | Vidrio deslustrado, nódulos centrolobulillares, engrosamiento septal y del intersticio peribroncovascular; adenopatías |
| Amiloidosis | Fina | Redondeados. A veces nódulos calcificados en su pared o en el interior | Difusa, peribroncovascular o subpleural | Nódulos múltiples, a menudo calcificados |
BHD: Birt-Hogg-Dubé; HCL: histiocitosis pulmonar de células de Langerhans; LLM: linfangioleiomiomatosis; NIL: neumonía intersticial linfoide.
Los principales mecanismos propuestos para explicar el desarrollo de quistes pulmonares son: 1) obstrucción bronquiolar parcial con mecanismo valvular: la infiltración de las paredes bronquiolares (por infiltrado linfocitario en la neumonía intersticial linfoide (NIL), por células musculares lisas en la linfangioleiomiomatosis, por células de Langerhans en la histiocitosis pulmonar, por amiloide) causaría dilatación/atrapamiento aéreo más distal7–12; 2) necrosis isquémica pulmonar focal, como la debida al depósito vascular de amiloide con reducción secundaria del flujo sanguíneo3,11,13,14; 3) destrucción de las paredes bronquiales o alveolares, como ocurre en la histiocitosis pulmonar por la infiltración de la pared de los bronquiolos por granulomas con células de Langerhans, o por infiltración de las paredes alveolares por células linfoplasmocíticas en el síndrome de Sjögren15,16; 4) acción de enzimas proteolíticas (metaloproteinasas) que degradan la matriz extracelular y las membranas elásticas de las vías aéreas, y se han relacionado con la aparición de quistes pulmonares en la histiocitosis de células de Langerhans, la linfangioleiomiomatosis y la enfermedad pulmonar por depósito de cadenas ligeras3,11,13,17, y 5) fibrosis retráctil con dilatación de espacios aéreos, como ocurre en la fase fibrótica de la histiocitosis de células de Langerhans5,6,15.
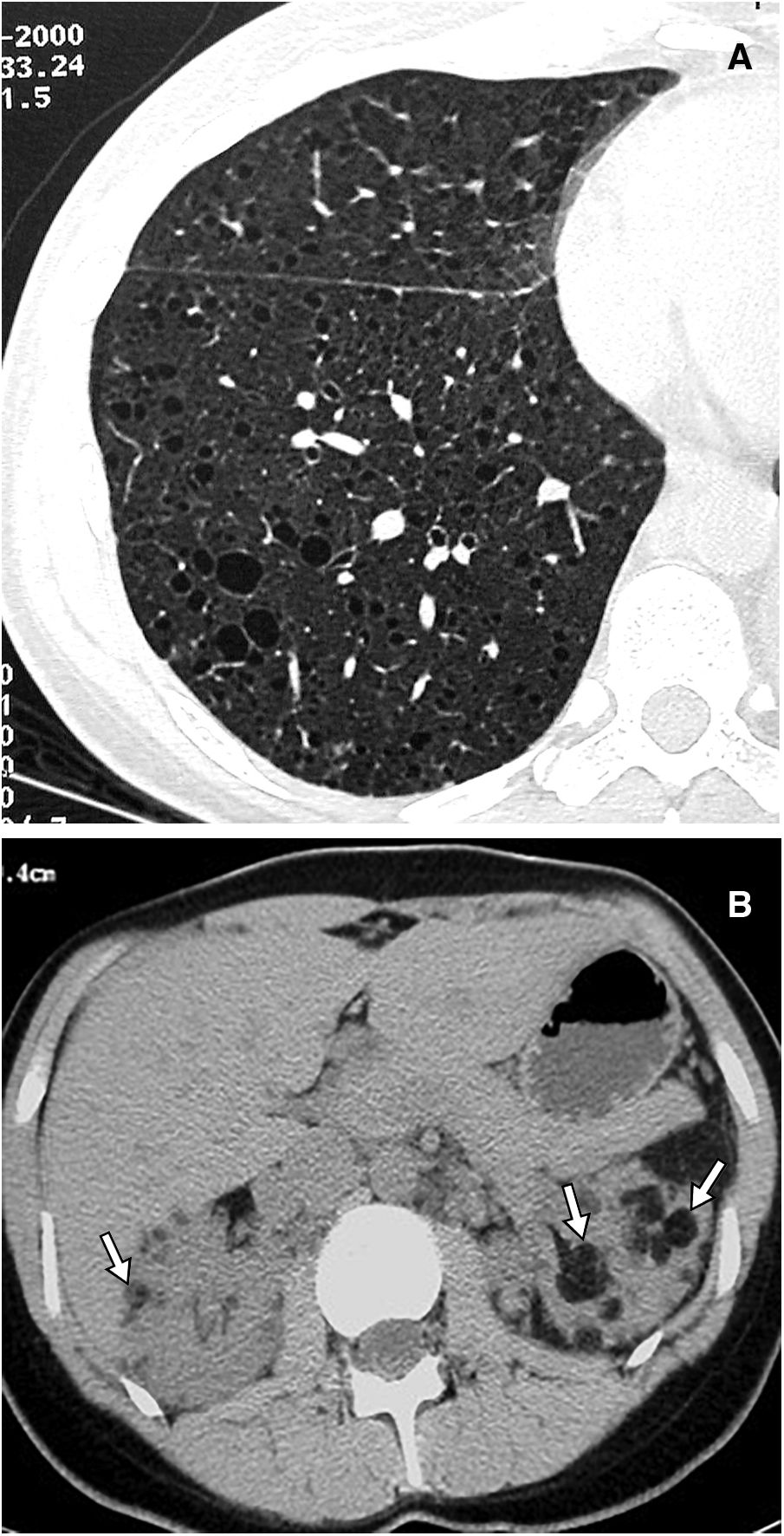
Principales enfermedades pulmonares quísticas difusasLinfangioleiomiomatosisLa linfangioleiomiomatosis (LLM) es una enfermedad sistémica infrecuente caracterizada por la infiltración del parénquima pulmonar por células musculares lisas anómalas, que conduce a su destrucción con formación de quistes pulmonares difusos. Se considera una neoplasia de bajo grado metastatizante, pero se desconoce el origen de esas células anómalas, que llegan al pulmón por vía linfática o sanguínea. En su patogenia están implicadas mutaciones en los genes TSC1 o TSC2, que codifican las proteínas hamartina y tuberina, y cuya alteración se traduce en una activación de la vía mTOR y secundariamente en una proliferación celular descontrolada, angiogénesis y linfangiogénesis. Hay formas esporádicas de LLM (mutaciones de TSC2 en células somáticas) y formas con herencia autosómica dominante asociadas al complejo de la esclerosis tuberosa (ET), que se caracteriza por retraso en el desarrollo, convulsiones, lesiones cutáneas y tumores benignos en múltiples órganos, con mutaciones de TSC1 y TSC2 en la línea germinal. Alrededor de un 30% de las mujeres y un 10% de los varones con ET tienen LLM. En casi todos los pacientes con LLM asociada a ET y en un 30-40% de los casos esporádicos hay angiomiolipomas renales (fig. 5) y pueden verse otras alteraciones linfáticas, como adenopatías, linfangioleiomiomas, dilatación u oclusión del conducto torácico, quilotórax o ascitis quilosa18,19.
Las formas esporádicas son exclusivas de mujeres y, aunque puede verse enfermedad quística pulmonar en un 10% de los varones con esclerosis tuberosa, la enfermedad pulmonar sintomática se da prácticamente solo en mujeres en edad fértil. Las células anómalas expresan receptores estrogénicos, lo que explica el empeoramiento más rápido de la función pulmonar en mujeres premenopáusicas, con el uso de estrógenos y durante el embarazo. La LLM es lentamente progresiva: a los 10 años del diagnóstico la mitad de las pacientes experimentan disnea con las actividades cotidianas, un 20% requiere oxígeno y un 10% ha fallecido. La supervivencia media es de más de 20 años tras el diagnóstico18,19.
Se ha propuesto como mecanismo de formación de los quistes una dilatación de los espacios aéreos distales por efecto valvular, secundaria a la estenosis de los bronquiolos terminales infiltrados por las células musculares anómalas5. Otro posible mecanismo implica a las metaloproteinasas, sobreexpresadas en las células de la LLM, que alterarían la matriz extracelular20,21. La infiltración por las células de la LLM también conduce a la destrucción de la pared arteriolar y oclusión de arteriolas pulmonares5.
Las formas de presentación más frecuentes de la LLM son el neumotórax recurrente en una mujer en edad fértil y la disnea progresiva, con un patrón obstructivo en la espirometría y restricción de la capacidad de difusión. El quilotórax es la manifestación inicial en un 20% de los casos. Otras presentaciones atípicas pueden ser abdominales, secundarias al sangrado de angiomiolipomas renales, al crecimiento de linfangioleiomiomas o a ascitis quilosa. Es cada vez más frecuente detectar la enfermedad incidentalmente en las pruebas de imagen realizadas por otro motivo, o puede encontrarse en el cribado de la esclerosis tuberosa.
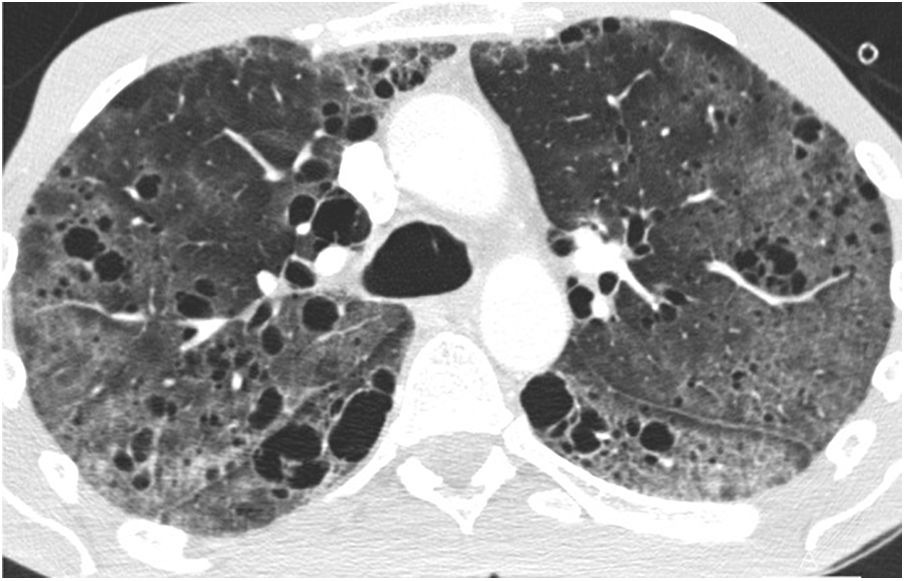
La radiografía de tórax en las fases iniciales suele ser normal y al progresar la enfermedad puede verse un aumento del volumen pulmonar con una afectación reticular o nodular; otros hallazgos son adenopatías, derrame o engrosamiento pleural. La TC es la prueba de imagen esencial para el diagnóstico de LLM y se recomienda para el cribado de LLM en mujeres en edad fértil con un primer episodio de neumotórax, en todas las mujeres con neumotórax de repetición y en pacientes con esclerosis tuberosa mayores de 18 años22–24. El principal hallazgo en la TC son los quistes aéreos de pared fina, de distribución difusa, con un tamaño desde pocos milímetros varios centímetros, redondeados, aunque de forma más atípica cuando crecen y confluyen13,14,25,26. En las fases iniciales los quistes son escasos y van aumentando en número y tamaño de manera que en las fases avanzadas queda poco parénquima normal entre ellos (figs. 5 y 6). Pueden verse vasos en la periferia de los quistes, pero no en su centro, lo que sirve para diferenciarlos del enfisema centroacinar. La presencia de quistes en los ángulos costofrénicos, su ausencia en las regiones apicales y la ausencia de nódulos, son datos útiles para el diagnóstico diferencial con la histiocitosis de células de Langerhans. Como pueden verse quistes pulmonares aislados como resultado del envejecimiento normal27, se ha marcado un mínimo de 4 quistes para considerarlos patológicos (y de 4 a 10 para diagnosticar LLM en pacientes con esclerosis tuberosa)18. Otros hallazgos infrecuentes son las opacidades en vidrio deslustrado (por proliferación de células musculares lisas, hemorragia alveolar o acúmulo de líquido linfático), el engrosamiento septal (por infiltración de linfáticos), o los nódulos centrolobulillares (por infiltración por células musculares lisas formando nódulos macroscópicos o por hiperplasia multifocal)18,28. También pueden verse adenopatías, derrame pleural o pericárdico, dilatación del conducto torácico, neumotórax (fig. 6) y linfangioleiomiomas quísticos.
 La imagen de TCAR centrada en el lóbulo inferior derecho muestra múltiples imágenes quísticas pulmonares. B) El corte de TC abdominal superior muestra múltiples angiomiolipomas renales bilaterales (flechas).")
Linfangioleiomiomatosis asociada a esclerosis tuberosa en una paciente mujer de 45 años. A) La imagen de TCAR centrada en el lóbulo inferior derecho muestra múltiples imágenes quísticas pulmonares. B) El corte de TC abdominal superior muestra múltiples angiomiolipomas renales bilaterales (flechas).
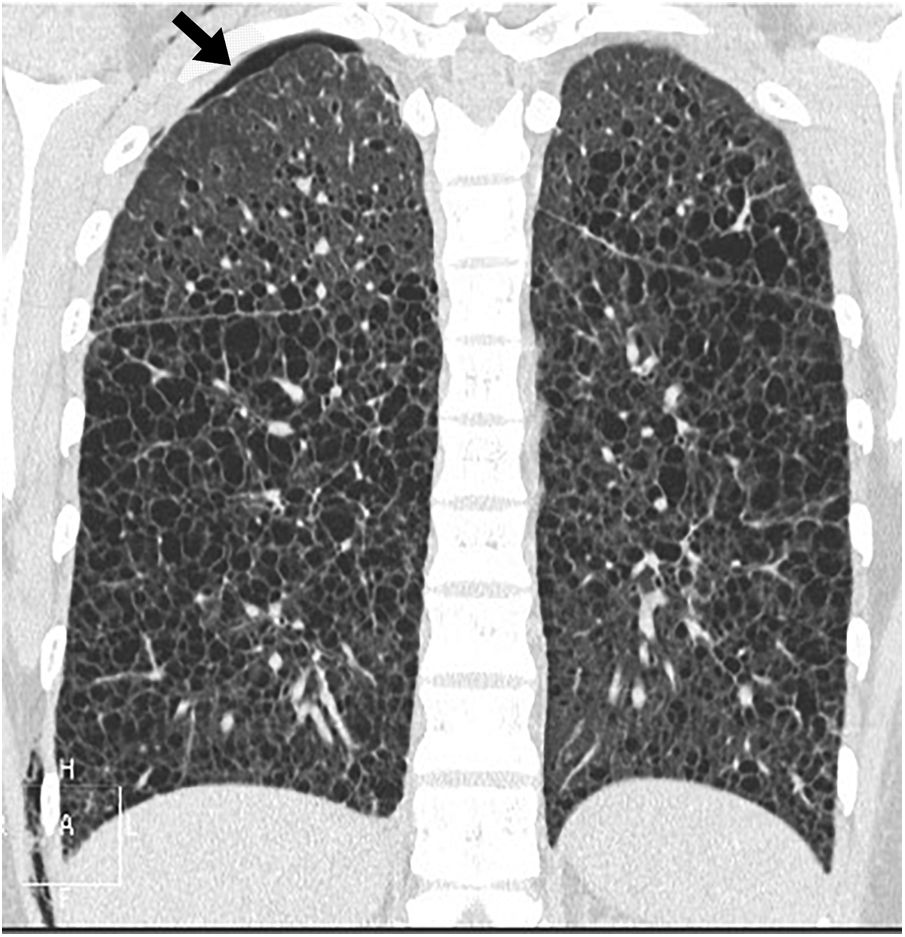
 y hay una pequeña cantidad de enfisema subcutáneo en la pared torácica lateral derecha.")
Linfangioleiomiomatosis en una mujer de 38 años. La imagen de reconstrucción en plano coronal muestra una extensa afectación quística pulmonar bilateral, con un discreto predominio en los campos pulmonares medios e inferiores. Destaca la presencia de un pequeño neumotórax apical derecho (flecha) y hay una pequeña cantidad de enfisema subcutáneo en la pared torácica lateral derecha.
La TC puede ser suficiente para establecer el diagnóstico de LLM en un contexto clínico adecuado (esclerosis tuberosa, angiomiolipomas renales, linfangiomas, quilotórax, ascitis quilosa, etc.). Las guías clínicas más recientes apoyan el uso de los niveles séricos del factor de crecimiento linfoangiogénico VEGF-D>800 pg/ml para establecer el diagnóstico en mujeres con una TC compatible, sin necesidad de biopsia29,30. La confirmación anatomopatológica se reserva para los casos con afectación pulmonar grave antes de iniciar el tratamiento con inhibidores mTORC1 (sirolimus, everolimus), que mejoran la función pulmonar. El trasplante pulmonar se plantea para las pacientes con enfermedad en fase muy avanzada, aunque se ha descrito la recidiva de la enfermedad en el pulmón trasplantado.
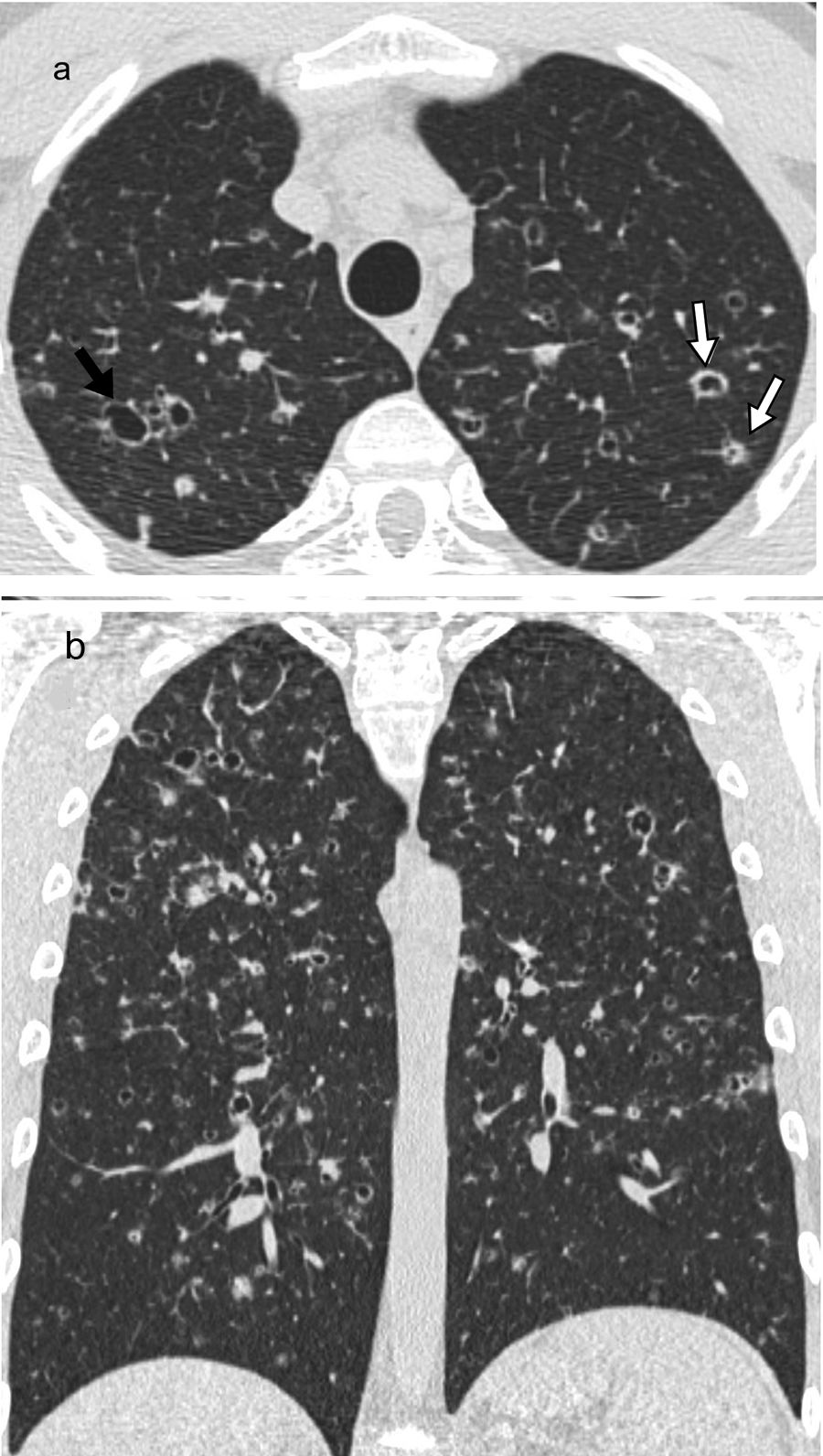
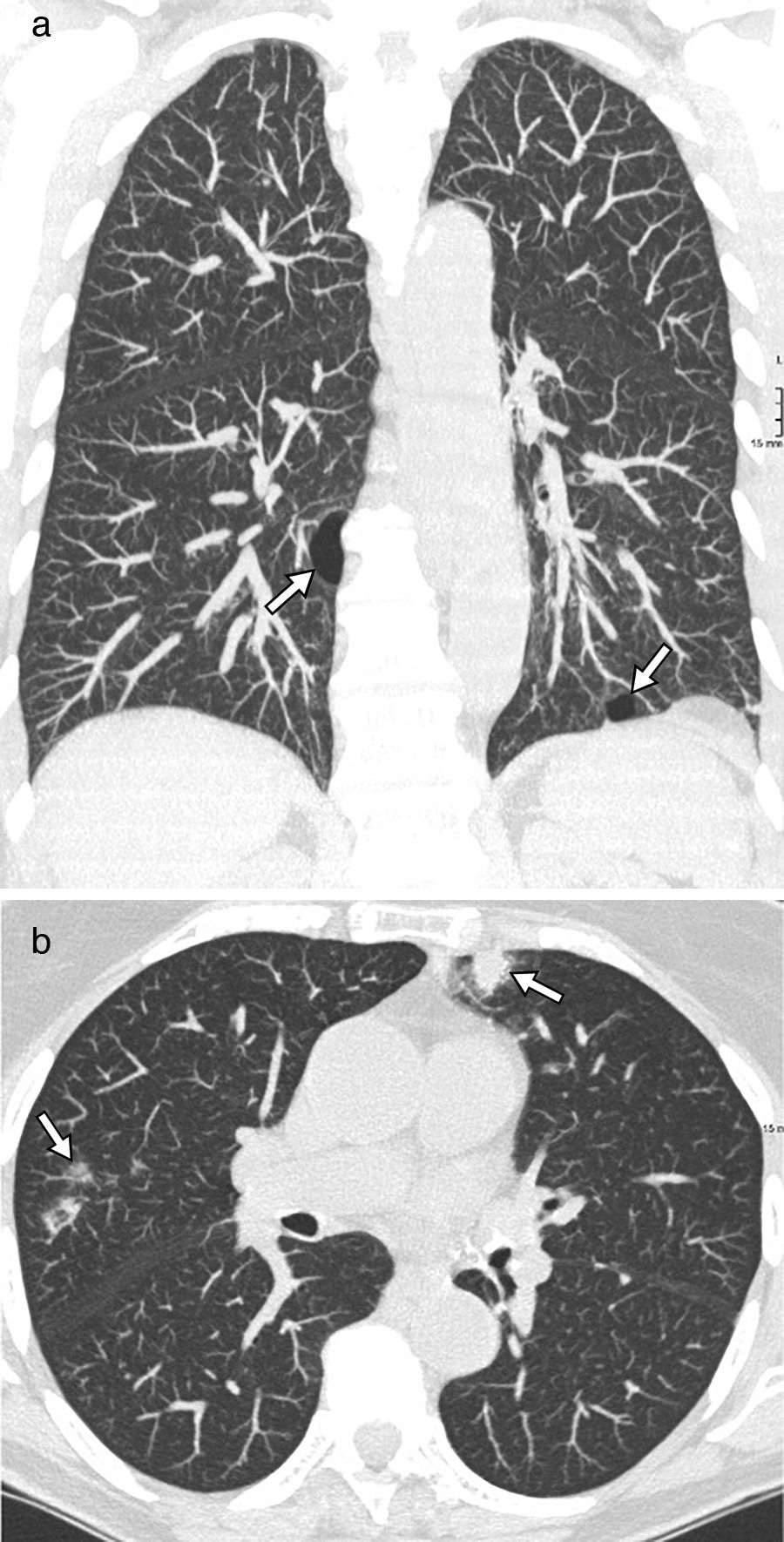
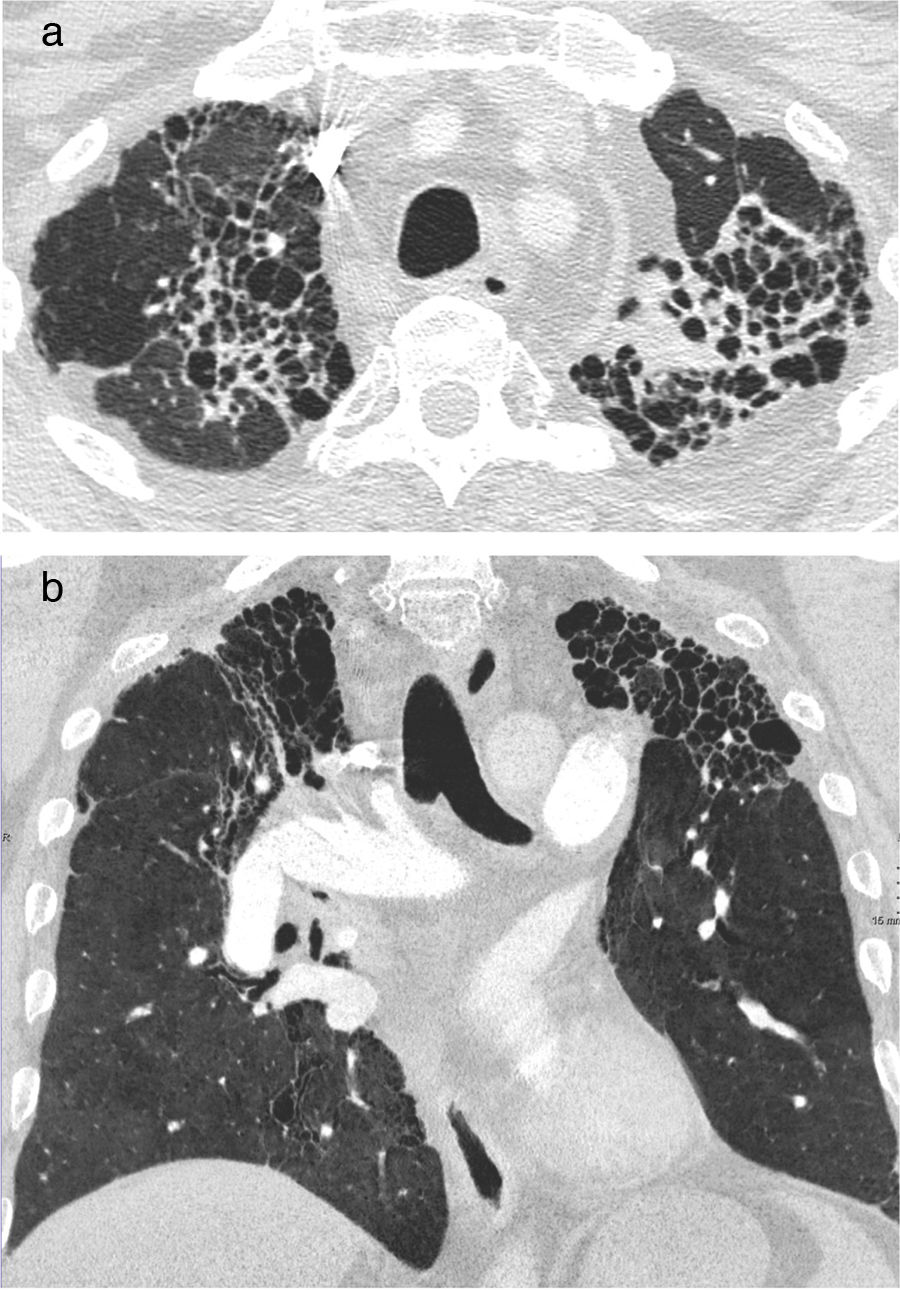
Histiocitosis de células de Langerhans (HCL)Es una enfermedad pulmonar intersticial infrecuente relacionada con el tabaco, que se diagnostica en adultos jóvenes con una historia actual o previa de tabaquismo. Se cree que componentes del humo del tabaco activan las células de Langerhans pulmonares que, junto con otras células inflamatorias, se acumulan formando infiltrados y granulomas peribronquiolares que se traducen en nódulos estrellados. Estos nódulos pueden mostrar cavidades que corresponden a dilatación bronquiolar. En las fases más avanzadas hay fibrosis peribronquial que tracciona y dilata los espacios aéreos contiguos14. Los síntomas más frecuentes son la disnea y la tos, pero a menudo no hay clínica respiratoria. Algunos pacientes inician con neumotórax (15%) y un pequeño porcentaje tienen síntomas sistémicos, dolores secundarios a afectación ósea, erupciones cutáneas o diabetes insípida. En las radiografías de tórax pueden verse nódulos en los lóbulos superiores en las fases iniciales y alteraciones reticulares y quísticas más adelante, con volúmenes pulmonares preservados o aumentados. En la TC en las fases iniciales predominan los nódulos centrolobulillares peribronquiolares, de 1-5mm, mal definidos o irregulares, con distribución bilateral y simétrica, de predominio en los campos superiores y medios, respetando los ángulos costofrénicos y la parte interna del lóbulo medio y la língula. Al progresar la enfermedad aparecen nódulos cavitados (fig. 7) con paredes gruesas y más adelante quistes de pared fina y diferentes tamaños, en general menores de 1cm, aunque en fases avanzadas pueden confluir y se ven quistes grandes y de morfología irregular o atípica (lobulados, ramificados, etc.) (fig. 8). Tanto anatomopatológicamente como en la TC hay heterogeneidad temporal y pueden coexistir nódulos cavitados con quistes de pared gruesa y fina2,5,6,12. El mecanismo sugerido de la formación de los quistes es la dilatación de los bronquiolos secundaria a la inflamación crónica con destrucción de su pared15; en las fases más avanzadas hay cicatrices fibrosas y enfisema paracicatricial peribronquiolar. La enfermedad puede regresar espontáneamente o tras dejar de fumar en un 25% de los pacientes, estabilizarse en un 50% o progresar a una enfermedad destructiva quística difusa en un 25% con insuficiencia respiratoria, incluso después de abandonar el tabaco. La hipertensión pulmonar es una complicación que se asocia a una mayor mortalidad y suele ser más grave que la asociada a otras causas, como el enfisema o la fibrosis pulmonar31. Su prevalencia es elevada, de alrededor del 40% en una serie reciente32, por lo que está indicado el cribado con ecocardiograma. En pacientes con una enfermedad de larga evolución y una importante afectación quística pulmonar, simulando un extenso enfisema, la existencia de signos de hipertensión pulmonar puede orientarnos al diagnóstico de una HCL. No es posible determinar con la TC el subgrupo de pacientes con HCL que va a progresar, por lo que es importante el seguimiento con pruebas de función respiratoria. El trasplante pulmonar es una opción en casos de enfermedad avanzada.
 La imagen de TCAR centrada en los lóbulos superiores muestra múltiples nódulos subcentimétricos de contornos irregulares, algunos cavitados (flechas blancas) y también imágenes quísticas aéreas (flecha negra). B) La imagen de reconstrucción en el plano coronal destaca la distribución predominante en los campos pulmonares superiores, con preservación de las bases pulmonares.")
Histiocitosis de células de Langerhans. A) La imagen de TCAR centrada en los lóbulos superiores muestra múltiples nódulos subcentimétricos de contornos irregulares, algunos cavitados (flechas blancas) y también imágenes quísticas aéreas (flecha negra). B) La imagen de reconstrucción en el plano coronal destaca la distribución predominante en los campos pulmonares superiores, con preservación de las bases pulmonares.
.")
El síndrome de Birt-Hogg-Dubé es una enfermedad multisistémica rara, transmitida por herencia autosómica dominante, causada por una mutación del gen de la foliculina en la línea germinal. Esta mutación puede ser heredada o aparecer de novo en pacientes sin historia familiar. Se manifiesta por lesiones cutáneas (fibrofoliculomas), múltiples quistes pulmonares, neumotórax espontáneos y tumores renales, que a menudo son multifocales, bilaterales y de lento crecimiento, con predominio de oncocitomas y neoplasias cromófobas. Hay una marcada variabilidad fenotípica: desde portadores asintomáticos a grados diversos de afectación pulmonar, cutánea y renal. Su prevalencia se estima en 1/200.000, pero su incidencia exacta se desconoce. Afecta a cualquier edad, sin predilección de género, aunque predomina a partir de la 3.ª-4.ª década33.
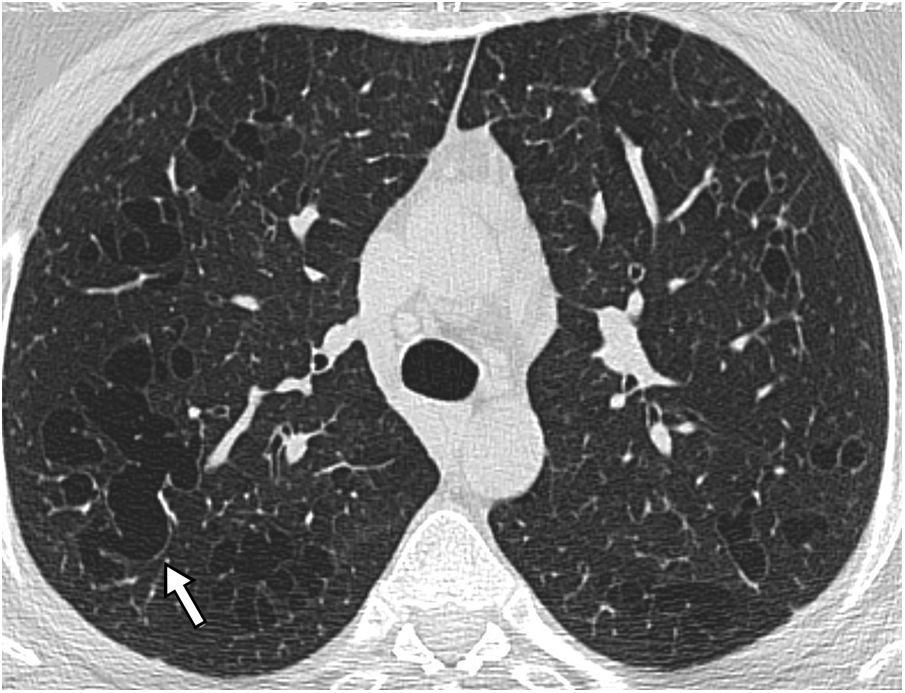
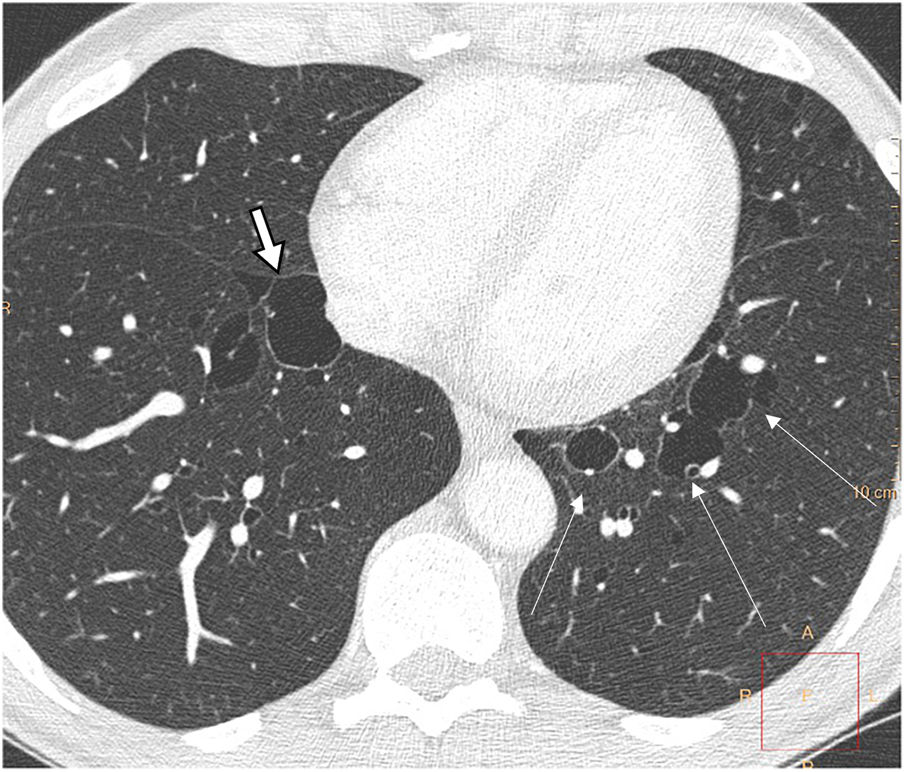
Los quistes pulmonares aparecen en el 80% de pacientes, en la juventud o edad media. En la TC tienen una pared fina, localización preferente en las bases, distribución periférica subpleural, incluyendo la localización paramediastínica y pericisural, y también en relación con septos interlobulillares, arterias y venas (fig. 9). Pueden ser redondos, pero también irregulares, elípticos y lenticulares, y los más grandes, septados. La presencia de quistes elípticos y paramediastínicos es especialmente indicativa de esta entidad y útil para el diagnóstico diferencial con otras enfermedades quísticas pulmonares. Los quistes son más grandes y menos numerosos que en la LLM y, a diferencia de esta, su número y tamaño no parece incrementarse progresivamente33–38. Microscópicamente están revestidos por neumocitos y parcialmente rodeados por tejido intersticial septal o pleural33,34.
 y otros adyacentes a vasos (flechas largas finas).")
Enfermedad de Birt-Hogg-Dubé en una mujer de 46 años. La imagen de TCAR a la altura de los lóbulos inferiores muestra múltiples imágenes quísticas aéreas de pared fina y morfología irregular, algunas de ellas en relación cisural (flecha) y otros adyacentes a vasos (flechas largas finas).
En el mecanismo de formación de los quistes se ha propuesto el papel de las alteraciones en la vía mTOR y de las metaloproteinasas33,34, que llevarían a la alteración de la matriz pulmonar, deficiente adhesión entre las células y expansión de los espacios alveolares como consecuencia del estrés inducido por los estiramientos en la respiración.
La mayoría de los pacientes con quistes pulmonares son asintomáticos, salvo en los episodios de neumotórax. A diferencia de la LLM y la histiocitosis, no experimentan deterioro progresivo de la función respiratoria. Los aspectos más importantes del manejo de estos pacientes son la pleurodesis tras el primer episodio de neumotórax espontáneo, por la alta incidencia de recidiva de neumotórax39, y el seguimiento para la detección precoz de tumores renales. Se debe hacer cribado de la enfermedad en familiares.
Neumonía intersticial linfoide/bronquiolitis folicularLa NIL es un trastorno linfoproliferativo benigno, caracterizado por la infiltración intersticial pulmonar por linfocitos policlonales, células plasmáticas e histiocitos, con predominio en el intersticio perilinfático (septal alveolar, interlobulillar, peribroncovascular y subpleural)40–43. Está incluida en la lista de las enfermedades intersticiales idiopáticas, aunque la mayoría de las veces es secundaria a otras enfermedades. La bronquiolitis folicular (BF) se caracteriza por la hiperplasia de folículos linfoides en las paredes de bronquiolos y vasos sanguíneos, aunque en algunos casos el infiltrado celular se extiende a los septos alveolares de forma focal, no difusa como en la NIL, lo que sugiere que estas entidades forman parte del mismo espectro de enfermedad11,14. Tanto la NIL como la BF se asocian a síndrome de Sjögren y otras enfermedades del colágeno (artritis reumatoide, lupus eritematoso sistémico), diversos trastornos autoinmunes, disproteinemias (hipergammaglobulinemia, hipogammaglobulinemia, inmunodeficiencia común variable), infección por virus de la inmunodeficiencia humana (sobre todo en niños) y otros virus40–43.
La NIL afecta a pacientes de edad media, con predominio en mujeres, y suele manifestarse por tos y disnea que progresan en meses o años, ocasionalmente con sintomatología sistémica asociada (fiebre, sudoración nocturna, pérdida de peso, artralgias). En las pruebas de función respiratoria hay un patrón restrictivo con una reducción de la capacidad de difusión.
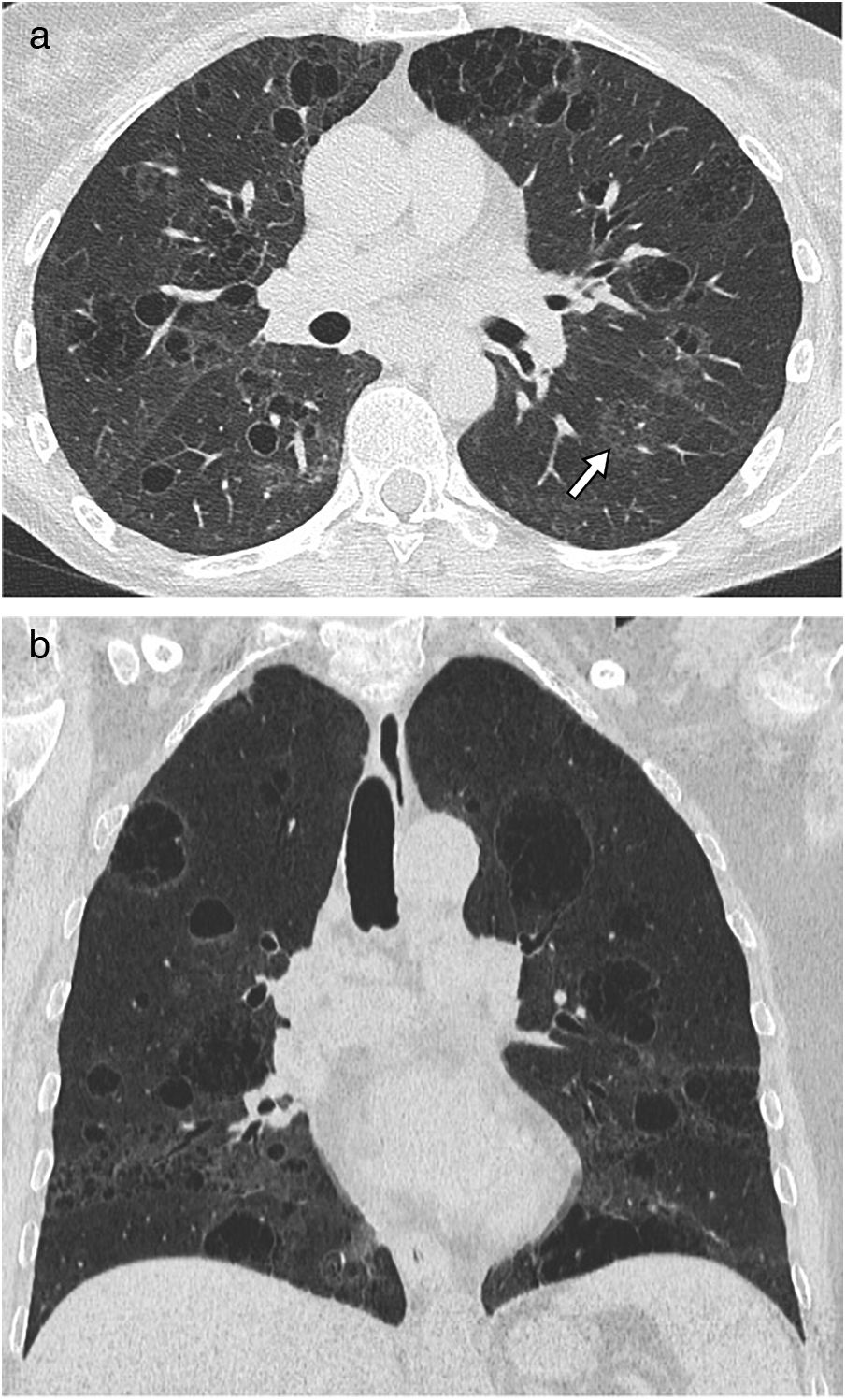
En las radiografías de tórax se ven infiltrados reticulonodulares y al progresar pueden aparecer opacidades en vidrio deslustrado y consolidaciones. Los hallazgos principales en la TC son las opacidades en vidrio deslustrado, los nódulos centrilobulillares y los quistes aéreos, presentes hasta en el 80% de los casos (fig. 10); también pueden verse nódulos subpleurales, engrosamiento septal y peribroncovascular, y adenopatías. Los quistes aéreos son poco numerosos, en general menores de 3cm, se ven con frecuencia en las zonas con vidrio deslustrado y tienen una distribución aleatoria, aunque a menudo se ven adyacentes a un vaso o un bronquio y también subpleurales44,45. En la TC de seguimiento pueden desaparecer los nódulos y el vidrio deslustrado, pero persistir los quistes46. Se ha propuesto como mecanismo patogénico de estos quistes la obstrucción parcial de la vía aérea secundaria a la infiltración linfocitaria peribronquiolar con atrapamiento aéreo distal; también puede haber isquemia por obstrucción vascular9,11,40,43–45. La identificación de estos quistes es útil para el diagnóstico diferencial con NINE celular y con linfoma, que puede ser difícil tanto en TC como en anatomía patológica40,42,43,47. En pacientes con NIL asociada a enfermedad de Sjögren la aparición de nódulos grandes debe hacer sospechar amiloidosis, especialmente si los nódulos están calcificados, o linfoma10,40 (fig. 11). La neumonía por hipersensibilidad puede presentar hallazgos muy similares a los de la NIL en la TC, con quistes en hasta un 13% de los casos8 y también puede ser difícil la diferenciación para el patólogo, pero habitualmente el contexto clínico ayuda al diagnóstico diferencial.
 en una mujer de 65 años con síndrome de Sjögren. A) La imagen de TCAR a la altura de los hilios pulmonares demuestra múltiples imágenes quísticas aéreas de paredes finas. Destaca también la presencia de opacidades en vidrio deslustrado (flecha). B) La imagen de reconstrucción en el plano coronal muestra la distribución difusa de los hallazgos.")
Neumonía intersticial linfoide (NIL) en una mujer de 65 años con síndrome de Sjögren. A) La imagen de TCAR a la altura de los hilios pulmonares demuestra múltiples imágenes quísticas aéreas de paredes finas. Destaca también la presencia de opacidades en vidrio deslustrado (flecha). B) La imagen de reconstrucción en el plano coronal muestra la distribución difusa de los hallazgos.
 asociada a linfoma MALT pulmonar en una mujer de 60 años con síndrome de Sjögren. La imagen de TCAR muestra quistes pulmonares y también nódulos pulmonares bilaterales de bordes irregulares y broncograma aéreo en su interior (flecha), correspondientes a focos de linfoma.")
Neumonía intersticial linfoide (NIL) asociada a linfoma MALT pulmonar en una mujer de 60 años con síndrome de Sjögren. La imagen de TCAR muestra quistes pulmonares y también nódulos pulmonares bilaterales de bordes irregulares y broncograma aéreo en su interior (flecha), correspondientes a focos de linfoma.
La evolución natural y el pronóstico son variables: pueden resolverse espontáneamente, mejorar o estabilizarse con corticoides o evolucionar a insuficiencia respiratoria y fibrosis pulmonar con aparición de panalización en la TC a pesar del tratamiento corticoideo40,46. Un 5% de los pacientes desarrolla un linfoma en el curso de la evolución. Alrededor del 30-50% de los pacientes fallece en 5 años.
Amiloidosis y enfermedad por depósito de cadenas ligerasEl término amiloidosis alude a un grupo heterogéneo de enfermedades que tienen en común el depósito extracelular de proteínas fibrilares insolubles con birrefringencia verde en la tinción con rojo Congo. La amiloidosis puede ser sistémica (80-90% de los casos) o localizada. La afectación pulmonar ocurre en alrededor del 50% de los casos y tiene 3 patrones fundamentales de distribución anatómica: traqueobronquial, enfermedad intersticial difusa y parenquimatosa nodular. La forma nodular pulmonar casi siempre es una enfermedad localizada, con depósito de amiloide AL, y forma parte del diagnóstico diferencial de las enfermedades quísticas pulmonares difusas. Se asocia a enfermedades del colágeno, fundamentalmente el síndrome de Sjögren, aunque también se da en pacientes sin conectivopatías, y a enfermedades linfoproliferativas, como el linfoma MALT10,16,48–52. En la TC se ven nódulos múltiples, de tamaño variable (de menos de 1cm a varios centímetros), de bordes lisos o espiculados, con distribución aleatoria o predominando en los campos inferiores, tanto peribroncovasculares como subpleurales, y con calcificación frecuente (hasta en 50%). Junto con los nódulos se ven quistes múltiples (fig. 12), de pared fina, distribución aleatoria, y a menudo se ven los nódulos calcificados en la pared del quiste o en su interior10,48–51. La posible patogenia de los quistes incluye la estenosis de la vía aérea por infiltración de la pared bronquiolar por amiloide y células inflamatorias, con dilatación distal por mecanismo valvular, la rotura de las paredes alveolares por fragilidad debida al depósito de amiloide e isquemia secundaria al depósito de amiloide en las paredes vasculares11,13,16,49. El diagnóstico diferencial, especialmente en pacientes con enfermedad de Sjögren, incluye la NIL, pero en esta los nódulos no calcifican y se ven nódulos centrolobulillares mal definidos.
 La imagen de reconstrucción MIP (maximum intensity projection) en el plano coronal muestra imágenes quísticas aéreas en ambos lóbulos inferiores (flechas). B) La imagen MIP en el plano transversal destaca la presencia de nódulos pulmonares bilaterales de contornos irregulares correspondientes a focos de amiloidosis (flechas).")
Amiloidosis pulmonar en forma quística y nodular. A) La imagen de reconstrucción MIP (maximum intensity projection) en el plano coronal muestra imágenes quísticas aéreas en ambos lóbulos inferiores (flechas). B) La imagen MIP en el plano transversal destaca la presencia de nódulos pulmonares bilaterales de contornos irregulares correspondientes a focos de amiloidosis (flechas).
La enfermedad pulmonar por depósito de cadenas ligeras es una entidad muy infrecuente, casi siempre asociada al mieloma múltiple o a la enfermedad de Waldenström. Se caracteriza por un depósito extracelular de material amorfo y un infiltrado linfoplasmocitario, sobre todo en el riñón, aunque también en la pared de los alvéolos y de la pequeña vía aérea. En la TC se pueden ver, como en la amiloidosis, quistes difusos de pared fina, nódulos de milímetros a varios centímetros, y adenopatías, pero a diferencia de la amiloidosis es poco frecuente que los nódulos calcifiquen, y no hay positividad del material extracelular en la tinción con rojo Congo2,10,53,54. Suele ser una enfermedad progresiva que conduce a insuficiencia respiratoria y puede requerir un trasplante pulmonar. Se ha implicado a las metaloproteasas en la destrucción tisular que conduce a la formación de quistes17.
Otras causas de quistes pulmonares difusosMetástasis pulmonares quísticas: pueden verse en pacientes con sarcomas y carcinomas epidermoides de cabeza y cuello, pero también con adenocarcinomas, con un predominio en los lóbulos inferiores (fig. 13).
.")
Infecciones: en la infección por Pneumocystis jiroveci el hallazgo dominante son las opacidades difusas en vidrio deslustrado y la reticulación, pero pueden verse lesiones quísticas aéreas hasta en un 35% de los pacientes (fig. 14). Tienen diferentes tamaños y grosor de pared, y una distribución difusa o con predominio en los lóbulos superiores; pueden resolverse al hacerlo la infección. La coccidiomicosis, enfermedad fúngica endémica en el suroeste de Estados Unidos, y países centro y sudamericanos, se manifiesta en las formas crónicas con nódulos cavitados, pero ocasionalmente persisten quistes de pared fina. En la equinococosis, la paragonimiasis y en las infecciones por micobacterias pueden verse también quistes pulmonares. La papilomatosis laringotraqueobronquial, causada por el virus del papiloma humano, puede presentar afectación pulmonar al cabo de más de 10 años del diagnóstico de la afectación traqueolaríngea y se caracteriza por nódulos pulmonares sólidos que evolucionan a lesiones quísticas redondeadas o irregulares de distribución difusa, con un grosor de pared variable, en ocasiones con niveles hidroaéreos (fig. 15).

 La imagen de TC de los campos superiores muestra un nódulo subcentimétrico en la pared traqueal correspondiente a un papiloma. B) La imagen de TC de los lóbulos inferiores muestra múltiples imágenes quísticas, algunas de ellas con un nivel hidroaéreo, y nodulares, correspondientes a una extensa afectación bilateral por papilomatosis.")
Papilomatosis traqueopulmonar en un paciente de 17 años. A) La imagen de TC de los campos superiores muestra un nódulo subcentimétrico en la pared traqueal correspondiente a un papiloma. B) La imagen de TC de los lóbulos inferiores muestra múltiples imágenes quísticas, algunas de ellas con un nivel hidroaéreo, y nodulares, correspondientes a una extensa afectación bilateral por papilomatosis.
Sarcoidosis: la sarcoidosis es una enfermedad granulomatosa multisistémica, caracterizada por la formación de granulomas no caseificantes en múltiples órganos. La afectación quística es rara, generalmente ocurre en pacientes con sarcoidosis fibrótica (estadio clínico IV) y se identifican quistes aglomerados que asemejan un patrón en panal (fig. 16). Se especula que estos quistes periféricos podrían ser atribuibles a un mecanismo valvular secundario a la fibrosis peribronquial o a la acumulación de granulomas55.
 En la imagen de TC de los lóbulos superiores destaca la presencia de quistes aéreos que simulan un patrón en panal. B) La imagen de reconstrucción coronal destaca la afectación predominante en los lóbulos superiores, con pérdida de volumen asociada.")
Sarcoidosis con afectación quística en un varón de 57 años. A) En la imagen de TC de los lóbulos superiores destaca la presencia de quistes aéreos que simulan un patrón en panal. B) La imagen de reconstrucción coronal destaca la afectación predominante en los lóbulos superiores, con pérdida de volumen asociada.
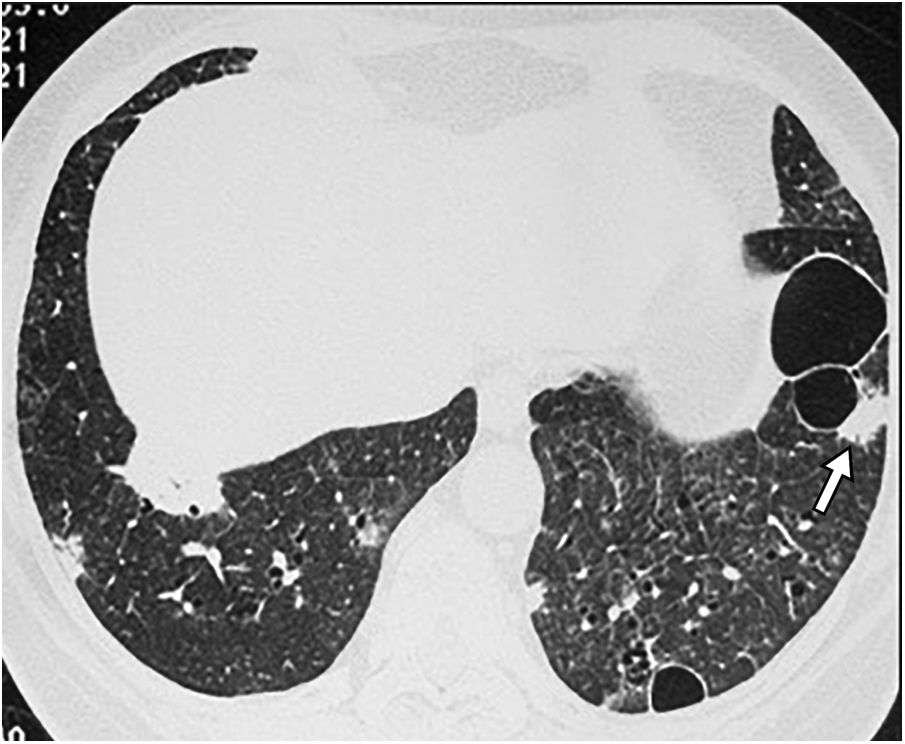
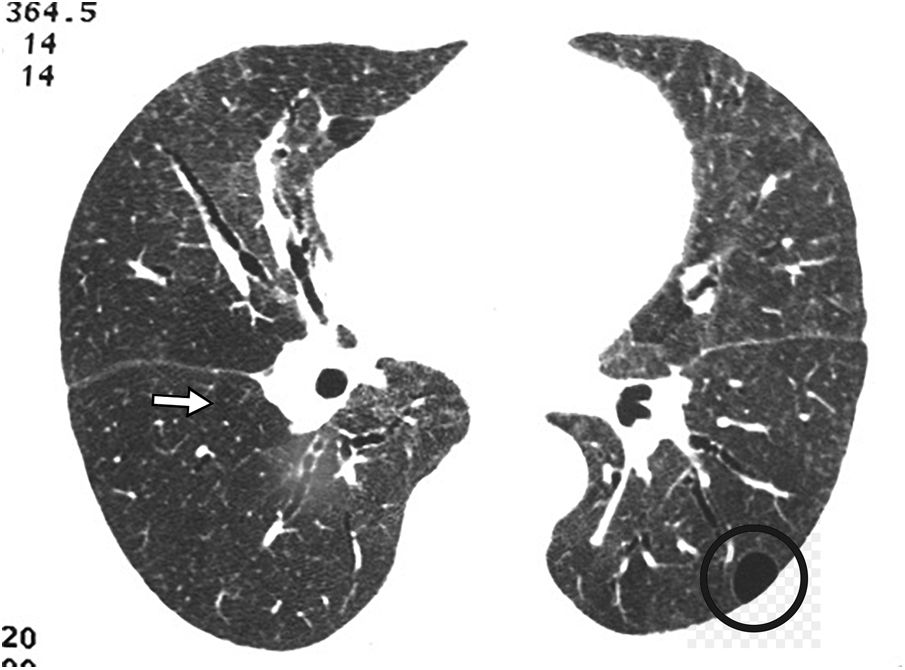
Neumonitis por hipersensibilidad (NH): la NH es una enfermedad difusa pulmonar causada por la inhalación de un amplio espectro de antígenos que desencadenan una reacción inmunitaria patológica en los pulmones en individuos predispuestos. A pesar de no ser una manifestación radiológica característica, en algunos casos presenta quistes aéreos pulmonares bilaterales, de distribución aleatoria56 (fig. 17).
Conclusiones y un quiste aéreo en el lóbulo inferior izquierdo (círculo).")
Las enfermedades quísticas pulmonares constituyen un grupo heterogéneo de entidades que se caracterizan por la presencia de lesiones aéreas de pared fina y distribución más o menos difusa. El diagnóstico diferencial puede ser complejo y el primer paso suele ser identificar la verdadera naturaleza quística de las lesiones, y también la distribución difusa de las mismas. Es de especial importancia analizar el tamaño, el número, la morfología y la distribución de los quistes, así como la presencia de otros hallazgos asociados. La TC de alta resolución (TCAR) constituye la técnica de imagen diagnóstica más útil en estas enfermedades y muchas veces hace innecesaria la confirmación histológica del diagnóstico.
Autoría1. Responsable de la integridad del estudio: BCM, AGP y SPMF.
2. Concepción del estudio: BCM, AGP y SPMF.
3. Diseño del estudio: BCM, AGP y SPMF.
4. Obtención de los datos: NA.
5. Análisis e interpretación de los datos: NA.
6. Tratamiento estadístico: NA.
7. Búsqueda bibliográfica: BCM, AGP y SPMF.
8. Redacción del trabajo: BCM, AGP y SPMF.
9. Revisión crítica del manuscrito con aportaciones intelectualmente relevantes: BCM, AGP y SPMF.
10. Aprobación de la versión final: BCM, AGP y SPMF.
Conflicto de interesesLos autores del artículo declaran no tener conflicto de intereses.

