




El feocromocitoma es un paraganglioma adrenal potencialmente maligno, con baja incidencia, pero relevancia evidente. Puede aparecer en varios síndromes hereditarios, especialmente von Hippel-Lindau, neoplasia endocrina múltiple 2 y paraganglioma familiar. En casos esporádicos subyacen también frecuentemente alteraciones genéticas que están cambiando el paradigma de la enfermedad.
Aunque puede tener una presentación clínica característica, en un 13,1-57,6% de los casos es el radiólogo el primero en sugerirlo, indicando determinaciones analíticas de catecolaminas o exploraciones de Medicina Nuclear.
Debe sospecharse ante una masa adrenal bien delimitada con realce intenso y rápido, mostrando característicamente fenómenos quísticos y hemorrágicos, alta señal en T2 y ausencia de lípidos macro o microscópicos. El comportamiento en difusión no suele aportar información muy relevante. Aproximadamente un tercio presentan lavado tardío similar al del adenoma en TC. Debe evitarse su punción percutánea ante su sospecha, por el riesgo de desencadenar una crisis hipertensiva grave.
Pheochromocytomas are adrenal paragangliomas. Potentially malignant, these tumors have a low incidence but clear importance. They can appear in various hereditary syndromes, especially in von Hippel-Lindau syndrome, multiple endocrine neoplasia-2 (MEN2), and familial paraganglioma syndromes. In sporadic cases, underlying genetic alterations are often found, and these findings are changing our understanding of the disease.
Although these tumors can manifest with a characteristic clinical presentation, in 13.1% to 57.6% of cases, it is the radiologist who first suggests the diagnosis, indicating analyses for catecholamines or nuclear medicine examinations.
Radiologists should suspect a pheochromocytoma on detection of a well-delimited adrenal mass with rapid, intense enhancement that typically shows cystic and hemorrhagic phenomena, high T2 signal intensity, and the absence of macroscopic or microscopic lipids. The behavior in diffusion-weighted imaging usually does not provide very useful information. Approximately one-third of lesions show late washout similar to that seen with adenomas on CT. Percutaneous puncture should be avoided to avoid the risk of unleashing a severe hypertensive crisis.
Los feocromocitomas se consideran, desde la actualización de la Organización Mundial de la Salud del 2022, paragangliomas adrenales potencialmente malignos1,2. Proceden de células precursoras de la médula adrenal, derivadas de la cresta neural, capaces de segregar varias catecolaminas, entre ellas, epinefrina.
Los paragangliomas extraadrenales (PGEA) pueden ser simpáticos (comúnmente abdominales y también secretores, aunque no de epinefrina) o parasimpáticos (la mayoría cervicales y no secretores)1–3. Los primeros pueden asentar en cualquier región de la cadena simpática retroperitoneal (especialmente en el órgano de Zuckerkandl, masa de células cromafines dispuestas en torno a la aorta abdominal y especialmente concentradas entre el origen de la arteria mesentérica inferior y la bifurcación aórtica), en el mediastino o en la vejiga, donde pueden generar episodios clínicos por liberación de catecolaminas durante la micción. El radiólogo tiene un papel fundamental en su diagnóstico y en la planificación quirúrgica.
Epidemiología y clínicaLos feocromocitomas son una de las causas potencialmente curables de hipertensión arterial (HTA), aunque se dan en menos del 0,2% de hipertensos. La incidencia anual es de 0,8/10.000, aunque probablemente están infradiagnosticados (en una serie de autopsias un 50% no se había manifestado clínicamente4). Se presentan sin diferencias significativas por sexo5. La edad media es de 47 años y el tamaño promedio en el momento del diagnóstico de 49mm.
Debe recordarse que es útil, aunque imprecisa, la nemotecnia del 10%: metastásicos (2-10% según las series; los PGEA hasta 20-25%), bilaterales, familiares (hasta un 40% en algunos estudios), extraadrenales y sin HTA.
Aproximadamente un 50% son sintomáticos, por secreción de catecolaminas, con síntomas paroxísticos de cefalea (90% de los pacientes sintomáticos), sudoración (60-70%) y taquicardia. Pueden inducir miocardiopatía similar al tako-tsubo. Esta sintomatología se acompaña de registros episódicos de HTA en la mitad de los casos y de HTA mantenida en 35-45%3,5–7. Un 5-15% no presentan HTA, porcentaje que está aumentando conforme también lo hacen los casos con diagnóstico incidental radiológico (13,1-57,6%).
En 2017 se incorporó su estadificación en la octava edición del Manual del American Joint Comittee on Cancer (tabla 1). Son predictores de riesgo metastásico: tamaño> 5cm, situación extraadrenal (doble riesgo de muerte) y mutación germinal en el gen de la subunidad B de succinato deshidrogenasa (SDHB) (generalmente en PGEA). Destacan 3escalas histopatológicas de riesgo de metástasis, con buen valor predictivo negativo1,2: Pheochromocytoma of the Adrenal gland Scaled Score (PASS), Grading system for Adrenal Pheochromocytoma (GAPP) y COmposite Pheochromocytoma/paragaglioma Prognostic Score (COPPS). Algunos estudios apuntan también a un comportamiento radiológico infiltrante e invasivo como dato predictor de malignidad.
Estadificación tumoral de feocromocitomas y paragangliomas según el American Joint Comittee on Cancer (AJCC)
| Tumor primario: T | Ganglios regionales. N | Metástasis a distancia: M | |
|---|---|---|---|
| X | No puede precisarse | No puede precisarse | |
| 0 | Sin metástasis en ganglios regionales | Sin metástasis a distancia | |
| 1 | <5cm, sin invasión extraadrenal | Con metástasis en ganglios regionales | Con metástasis a distancia• M1a: a hueso• M1b: a ganglios no regionales, hígado o pulmón.• M1c: a hueso y otros órganos |
| 2 | ≥ 5cm, sin invasión extraadrenal o PGEA simpático de cualquier tamaño | ||
| 3 | Cualquier tamaño, con invasión extraadrenal |
Estadio I: T1 N0 M0 (en blanco). Estadio II: T2 N0 M0 (gris claro). Estadio III: T1-2 N1 M0, T3 N0-1 M0 (gris intermedio). Estadio IV: T1-2-3 N0-1 M1 (gris oscuro).
Modificado de AJCC Cancer Staging Manual, 8th edition. Springer, 2017.
Las metástasis más habituales se producen en los ganglios regionales, el hueso, el hígado y el pulmón3,6–8.
Feocromocitoma y genéticaEntre el 10 y el 40% de los pacientes tienen feocromocitomas o PGEA dentro de un síndrome hereditario (tabla 2). Suelen ser más jóvenes, con lesiones más pequeñas y homogéneas, con mayor frecuencia bilaterales9–11. Destacan: von Hippel-Lindau (fig. 1), neoplasia endocrina múltiple 2 (MEN 2) (fig. 2), paraganglioma hereditario y neurofibromatosis 1.
Feocromocitomas y PGEA en síndromes hereditarios
| Genes implicados (y loci) | Tipo herencia | Epidemiología | % FC y PGEA | % aprox. malignidad | Otros datos de interés | Otras entidades asociadas al síndrome | |
|---|---|---|---|---|---|---|---|
| Von Hipple-Lindau | VHL (3p25-26) | AD, alta penetrancia | 1/36.000-50.000 nacidos vivos | 25-30% FC | <5% | Tipo 1. Bajo riesgo FC | HB cerebelosos |
| FC a 27 años de edad media | 15% PGEA | Tipo 2A. Alto riesgo FC y bajo CRCC | HB medulares | ||||
| 20-50% FC bilateral | Tipo 2B. Alto riesgo FC y CRCC | CRCC | |||||
| Tipo 2C. Alto riesgo FC solo | Quistes renales | ||||||
| TNE pancreáticos | |||||||
| Quistes pancreáticos | |||||||
| CAS pancreáticos | |||||||
| Tumores del saco endolinfático | |||||||
| Cistoadenomas papilares epidídimo | |||||||
| MEN 2 | RET (10q11.21) | AD, muy alta penetrancia | Prevalencia 1/30.000 | 40-50% FC | 5% | MEN 2A: formas clásicas, con amiloidosis asociada a liquen cutáneo y con enfermedad de Hirchsprung, todas con incidencia similar de FC | MEN 2A: |
| MEN 2B: 6% de MEN 2 | Infrecuente PGEA | Carcinoma medular de tiroides (> 90%) | |||||
| 30-100% FC bilateral | Hiperplasia paratiroidea con hiperparatiroidismo (10-20%) | ||||||
| MEN 2B: | |||||||
| Carcinoma medular de tiroides (> 90%, más agresivo) | |||||||
| Neuromas mucosos. | |||||||
| Ganglioneuromas intestinales | |||||||
| Aganglionosis colónica | |||||||
| Hábito marfanoide | |||||||
| Mielinización de nervios corneales. | |||||||
| PGL hereditarioa | PGL 1: SDHD (11q23). | AD, penetrancia intermedia | Infrecuentes. Incidencia real desconocida | PGL 1: FC y PGEA S 40%, PGEA PS> 80% | PGL 1: <2% | La nomenclatura PGL 1-5 tiende a sustituirse por la alteración genética (MAX, SDHA, SDHAF2, SDHB, SDHC, SDHD y TMEM127) | GIST (sobre todo gástricos) |
| PGL 4: SDHB (11p35) | 74% tumores bilaterales | PGL 4: PGEA S> 80% | PGL 4: 35-70% | Condromas pulmonares | |||
| CRCC | |||||||
| Otros | |||||||
| NF1 | NF1 (17q11.2) | AD, penetrancia completa | 1/2.600-3.000 nacidos vivos | 3% FC | 12% | Es excepcional que haya FC bilaterales o PGEA | Manchas cutáneas café con leche |
| 50% no heredado | Pecas inguino-axilares | ||||||
| Nódulos de Lisch iridianos | |||||||
| Neurofibromas cutáneos y neurales | |||||||
| Neurofibromas plexiformes y nodulares | |||||||
| Gliomas ópticos y otros | |||||||
| Sarcomas de partes blandas | |||||||
| Tumores malignos de la vaina nerviosa | |||||||
| GIST | |||||||
| Tumores glómicos | |||||||
| Otros tumores benignos y malignos | |||||||
| Displasias óseas y seudoartrosis | |||||||
| Otras lesiones óseas | |||||||
| Déficit cognitivo | |||||||
| Macrocefalia | |||||||
| Cardiopatías congénitas | |||||||
| Hipertensión arterial | |||||||
| Otras |
AD: autosómica dominante; CAS: cistoadenomas serosos; CRCC: carcinomas renales de células claras; HB: hemangioblastomas; FC: feocromocitoma; GIST: tumor del estroma gastrointestinal; MEN 2: neoplasia endocrina múltiple 2; NF1: neurofibromatosis 1; PGEA: paraganglioma extraadrenal; PGL: paraganglioma; PS: parasimpático; S: simpático; TNE: tumores neuroendocrinos.
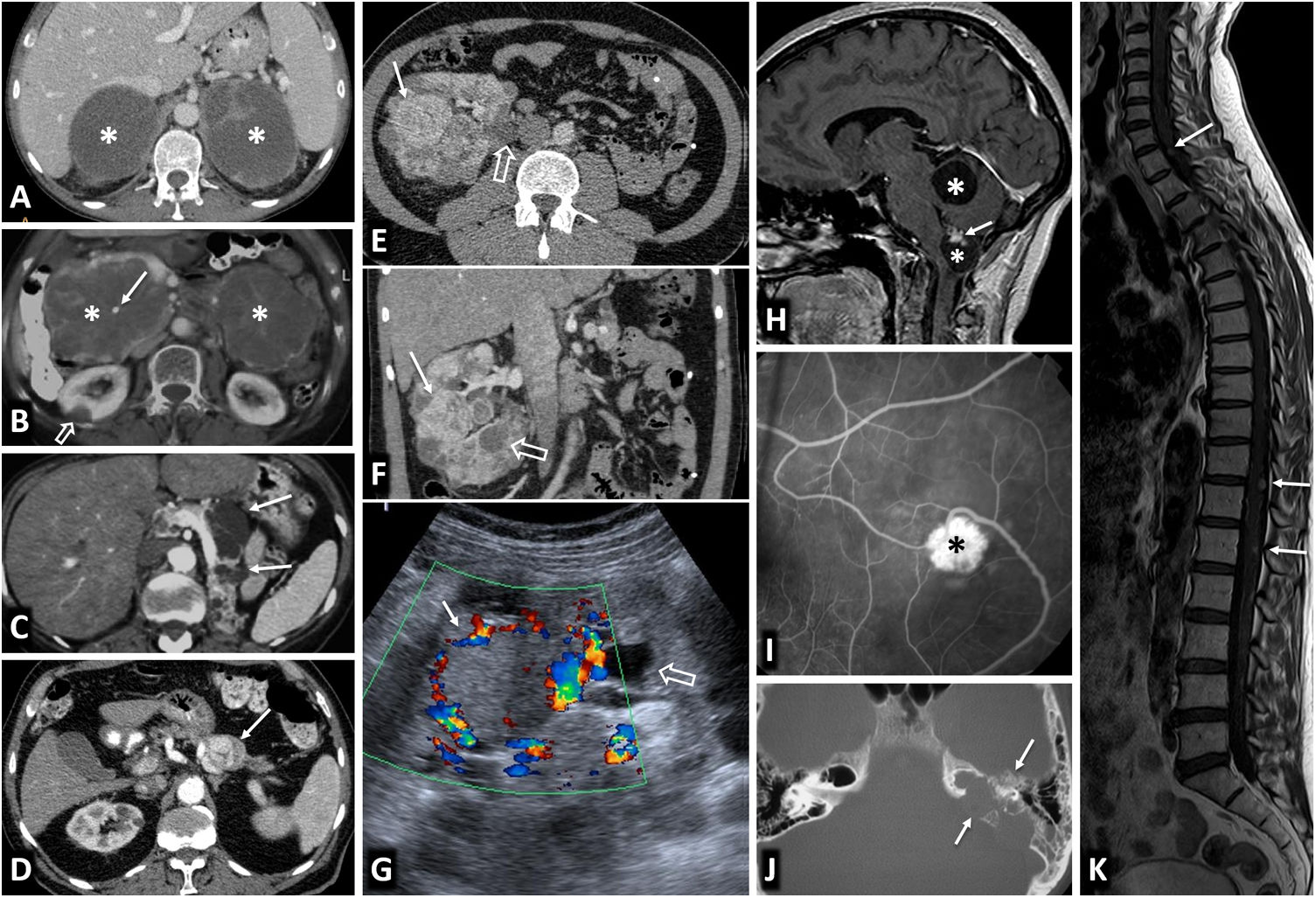
![Síndrome de von Hippel-Lindau. Hallazgos en distintos pacientes. A) Feocromocitomas adrenales bilaterales en un varón joven. TC: grandes masas bien delimitadas en ambas adrenales (*), con áreas quísticas predominantes. B) Cistoadenomas serosos pancreáticos múltiples en una mujer joven. TC: se ven 2 grandes masas pancreáticas (*) multiquísticas, con septos radiantes y, en la derecha, una pequeña calcificación central (flecha). También se ve un quiste renal cortical derecho (flecha hueca). C) Quistes pancreáticos múltiples. TC: quistes de diversos tamaños (flechas) en el cuerpo y la cola de un páncreas reposicionado tras nefrectomía izquierda por múltiples carcinomas renales de células claras. D) Tumor neuroendocrino pancreático. TC: pequeña lesión nodular (flecha) con intenso realce en la fase arterial, en el cuerpo pancreático. También se ven pequeños quistes renales. E, F [TC] y G [ecografía Doppler]) Múltiples carcinomas de células claras (flechas) y quistes corticales (flechas huecas) en el riñón derecho único de un varón joven ya sometido a nefrectomía izquierda. H). Dos hemangioblastomas cerebelosos. RM sagital T1 con Gd: lesiones quísticas cerebelosas (*), con un nódulo sólido con realce heterogéneo en la más inferior (flecha). I) Angioma retiniano. Angiograma retiniano con fluoresceína. Lesión hiperfluorescente (*) con un aporte arterial prominente. J) Tumor del saco endolinfático. TC: lesión lítica (flechas) con su epicentro en la cresta petrosa posterior en la región del acueducto vestibular, con algunas espículas óseas intratumorales. K) Hemangioblastomas espinales múltiples. RM sagital T1 con Gd: pequeños focos de realce intramedular (flechas).](https://static.elsevier.es/multimedia/00338338/0000006400000004/v1_202207191650/S0033833822001400/v1_202207191650/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNfYM26a8Bai41R8kcJS5RRAf6ffZrG0r8IP2HfWtJ9XVXZ5z2r3pCrQW8lORcm8QNromXdu4rQQ4FaRKz0WspbNuYX3l184ECPG+HFB5GlIHVWXvwU57i21MGvx4dm6meURZSNbXDXXMS86SUnhipUDDu/CqDPxOm4UMUIjdB3cZ95rgZ7mOTfqBrSsNJCI6TLq7Fgv3wx6fxVGmDvFNBTTh0+jbfFjW3lx7ZZqEfujT9BXyNIyowXDySqjx4sa5QM= "Síndrome de von Hippel-Lindau. Hallazgos en distintos pacientes. A) Feocromocitomas adrenales bilaterales en un varón joven. TC: grandes masas bien delimitadas en ambas adrenales (*), con áreas quísticas predominantes. B) Cistoadenomas serosos pancreáticos múltiples en una mujer joven. TC: se ven 2 grandes masas pancreáticas (*) multiquísticas, con septos radiantes y, en la derecha, una pequeña calcificación central (flecha). También se ve un quiste renal cortical derecho (flecha hueca). C) Quistes pancreáticos múltiples. TC: quistes de diversos tamaños (flechas) en el cuerpo y la cola de un páncreas reposicionado tras nefrectomía izquierda por múltiples carcinomas renales de células claras. D) Tumor neuroendocrino pancreático. TC: pequeña lesión nodular (flecha) con intenso realce en la fase arterial, en el cuerpo pancreático. También se ven pequeños quistes renales. E, F [TC] y G [ecografía Doppler]) Múltiples carcinomas de células claras (flechas) y quistes corticales (flechas huecas) en el riñón derecho único de un varón joven ya sometido a nefrectomía izquierda. H). Dos hemangioblastomas cerebelosos. RM sagital T1 con Gd: lesiones quísticas cerebelosas (*), con un nódulo sólido con realce heterogéneo en la más inferior (flecha). I) Angioma retiniano. Angiograma retiniano con fluoresceína. Lesión hiperfluorescente (*) con un aporte arterial prominente. J) Tumor del saco endolinfático. TC: lesión lítica (flechas) con su epicentro en la cresta petrosa posterior en la región del acueducto vestibular, con algunas espículas óseas intratumorales. K) Hemangioblastomas espinales múltiples. RM sagital T1 con Gd: pequeños focos de realce intramedular (flechas).")
Síndrome de von Hippel-Lindau. Hallazgos en distintos pacientes. A) Feocromocitomas adrenales bilaterales en un varón joven. TC: grandes masas bien delimitadas en ambas adrenales (*), con áreas quísticas predominantes. B) Cistoadenomas serosos pancreáticos múltiples en una mujer joven. TC: se ven 2 grandes masas pancreáticas (*) multiquísticas, con septos radiantes y, en la derecha, una pequeña calcificación central (flecha). También se ve un quiste renal cortical derecho (flecha hueca). C) Quistes pancreáticos múltiples. TC: quistes de diversos tamaños (flechas) en el cuerpo y la cola de un páncreas reposicionado tras nefrectomía izquierda por múltiples carcinomas renales de células claras. D) Tumor neuroendocrino pancreático. TC: pequeña lesión nodular (flecha) con intenso realce en la fase arterial, en el cuerpo pancreático. También se ven pequeños quistes renales. E, F [TC] y G [ecografía Doppler]) Múltiples carcinomas de células claras (flechas) y quistes corticales (flechas huecas) en el riñón derecho único de un varón joven ya sometido a nefrectomía izquierda. H). Dos hemangioblastomas cerebelosos. RM sagital T1 con Gd: lesiones quísticas cerebelosas (*), con un nódulo sólido con realce heterogéneo en la más inferior (flecha). I) Angioma retiniano. Angiograma retiniano con fluoresceína. Lesión hiperfluorescente (*) con un aporte arterial prominente. J) Tumor del saco endolinfático. TC: lesión lítica (flechas) con su epicentro en la cresta petrosa posterior en la región del acueducto vestibular, con algunas espículas óseas intratumorales. K) Hemangioblastomas espinales múltiples. RM sagital T1 con Gd: pequeños focos de realce intramedular (flechas).
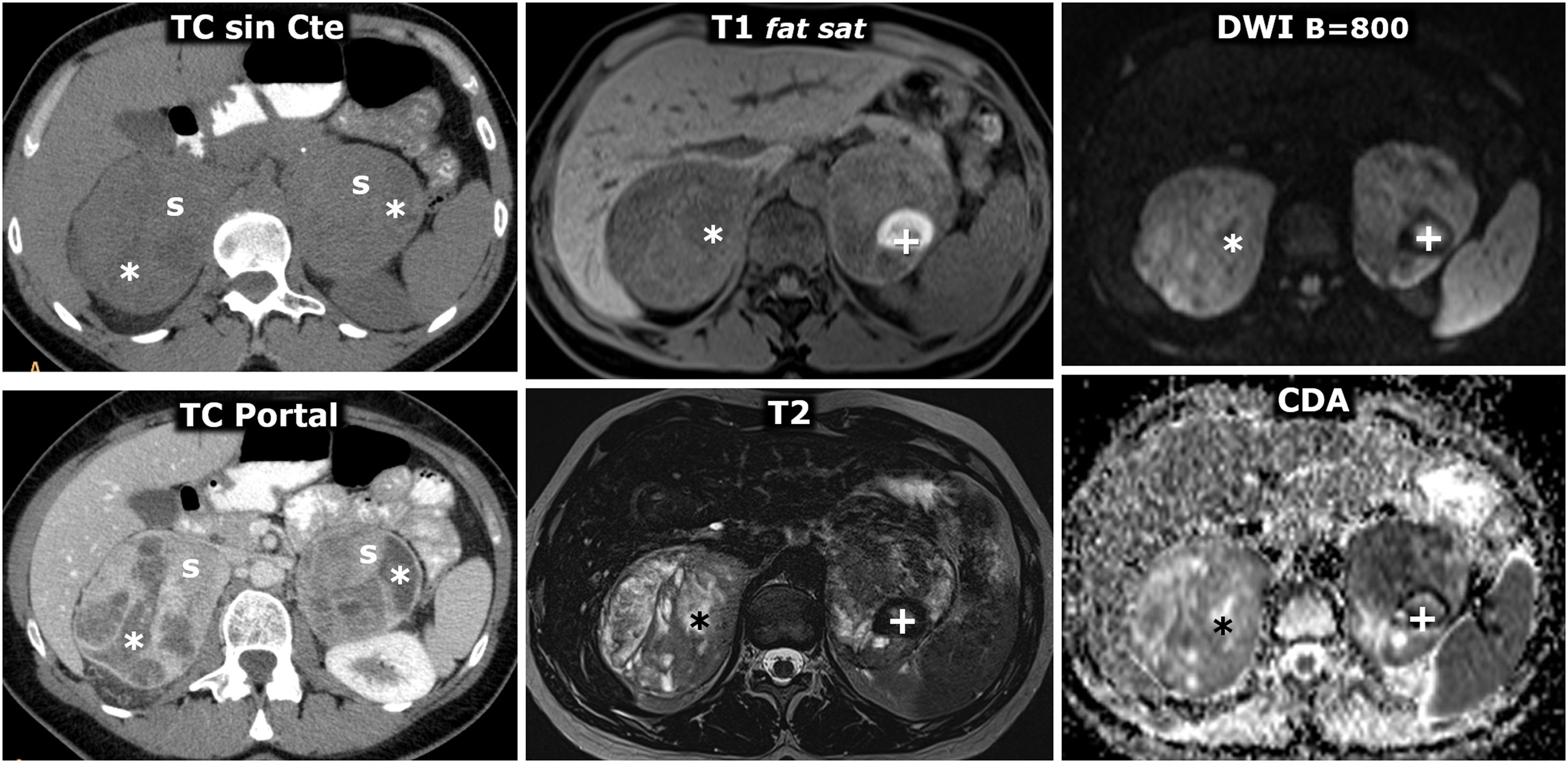
. Mujer de 42 años que consulta por episodios de hipoglucemia. Columna a la izquierda, TC: 2 masas adrenales grandes y bien delimitadas, con áreas sólidas (s) que realzan de forma intensa, y otras quísticas (*), sin realce significativo. Dos columnas a la derecha, RM: sendas masas adrenales, con algunos focos de señal especialmente alta en T2, que corresponden a focos de degeneración quística (*) y un área con alta señal periférica en T1, con muy baja señal en T2 (+), que se corresponde con un foco de hemorragia antigua con depósito periférico de hemosiderina.")
Síndrome MEN2A con feocromocitoma bilateral y carcinoma medular de tiroides milimétrico (no mostrado, 2 focos identificados en tiroidectomía total profiláctica). Mujer de 42 años que consulta por episodios de hipoglucemia. Columna a la izquierda, TC: 2 masas adrenales grandes y bien delimitadas, con áreas sólidas (s) que realzan de forma intensa, y otras quísticas (*), sin realce significativo. Dos columnas a la derecha, RM: sendas masas adrenales, con algunos focos de señal especialmente alta en T2, que corresponden a focos de degeneración quística (*) y un área con alta señal periférica en T1, con muy baja señal en T2 (+), que se corresponde con un foco de hemorragia antigua con depósito periférico de hemosiderina.
Entre un 24 y un 70% de los pacientes con feocromocitoma-PGEA considerado esporádico (incidental o no) presentan alguna anomalía genética (tabla 3) en las líneas de seudohipoxia, que implica una activación inapropiada del factor inductor de hipoxia (clústeres 1A-1B), de señalización de la cinasa, con estimulación de vías tumorales PI3K o RAS (clúster 2, más frecuente en feocromocitoma y asociado a menor agresividad) o de señalización Wnt, que implica hipometilación de ADN y mal pronóstico (clúster 3), con muchas consecuencias diagnósticas y terapéuticas conocidas y por definir8,10–13.
Alteraciones genéticas descritas en feocromocitomas y paragangliomas extraadrenales según el Atlas del Genoma del Cáncer
| Vía implicada | % estimado | Genes | Tipo de tumor | Riesgo de metástasis % | Otros síndromes y tumores | |
|---|---|---|---|---|---|---|
| Clúster 1A (VHL/EPAS1) | Pseudohipoxia (activación de factor inductor de hipoxia) | 15-20 | VHL | FC>>PAGEA | 5 | VHL |
| EPAS1/HIF2α | FC/PGTA | 29 | PC, Somatostatinoma | |||
| EGLN1,2 | FC/PGTA | ¿? | PC | |||
| Clúster 1B | 10-15 | SDHx | PGEA | Bajo | GIST, CCR, AdHf | |
| (TCA) | SDHA | PGTA>>PGCC/FC | 30-70 | GIST, CCR, AdHf | ||
| SDHB | PGCC | Bajo | GIST, CCR | |||
| SDHC | PGEA>FC | <5 | GIST, CCR, AdHf | |||
| SDHD | PGCC | Bajo | Leiomioma, CCR | |||
| SDHAF2 | FC/PGEA | >50 | Glioma bajo grado | |||
| FH | PGTA | ¿? | LMA | |||
| MDH2 | PGEA | ¿? | LMA | |||
| IDH1/IDH2 | PGEA | ¿Alto? | ||||
| SLC25A11 | PGCC | ¿? | ||||
| IDH3B | PGTA | ¿Alto? | ||||
| GOT2 | PGCC | ¿? | ||||
| DNMT3A | FC/PGEA | ¿? | ||||
| DLST | ||||||
| Clúster 2 | Señalización Kinasa | 50-60 | RET | FC | <5 | MEN 2 |
| NF1 | FC | 12 | NF1 | |||
| TMEM 127 | FC>PGEA | Bajo | CCR | |||
| MAX | FC/PGEA | 10 | Onconcitoma renal | |||
| H-RAS | FC | Bajo | Neuroblastoma | |||
| KIF1B | FC | ¿? | MEN 1 | |||
| MEN1 | FC/PGCC | ¿? | ||||
| Clúster 3 | Señalización Wnt | 5-10 | MAML3 | FC/PGCC | Alto | Neuroblastoma |
| CSDE1 | FC/PGCC | Alto |
Recientemente se ha descrito un cuarto clúster, aún poco definido. Además, se han descrito relaciones con otros genes habitualmente implicados en tumores, como TP53 y BRAF.
AdHf: adenoma hipofisario; CCR: carcinoma de células renales; FC: feocromocitoma; GIST: tumor del estroma gastrointestinal; LMA: leucemia mieloide aguda; MEN: neoplasia endocrina múltiple; PC: policitemia; PGEA: paraganglioma extraadrenal; PGCC: paraganglioma de cabeza y cuello; PGTA: paraganglioma torácico o abdominal; TCA: ácido tricarboxílico; VHL: von Hippel-Lindau.
Adaptado de Koopman et al.2.
Una sintomatología compatible, con o sin HTA, el diagnóstico de HTA a edad temprana o una historia familiar orientativa son las principales situaciones que justifican el estudio bioquímico o genético. Los matices diagnósticos están disponibles en guías y los artículos de referencia3,6–8,14–16.
Tras la confirmación bioquímica o ante alta sospecha clínica debe realizarse inicialmente TC o RM, según las peculiaridades del caso y el medio.
Debe recordarse que la punción percutánea está contraindicada, por el riesgo de desencadenar una crisis por descarga de catecolaminas.
Los estudios de Medicina Nuclear se emplean cuando los resultados de la TC y la RM son negativos persistiendo alta sospecha o en casos dudosos17–19. Destacan:
- –
(123I) MIBG SPECT, con precisión diagnóstica del 95%, superior a la gammagrafía (fig. 3).
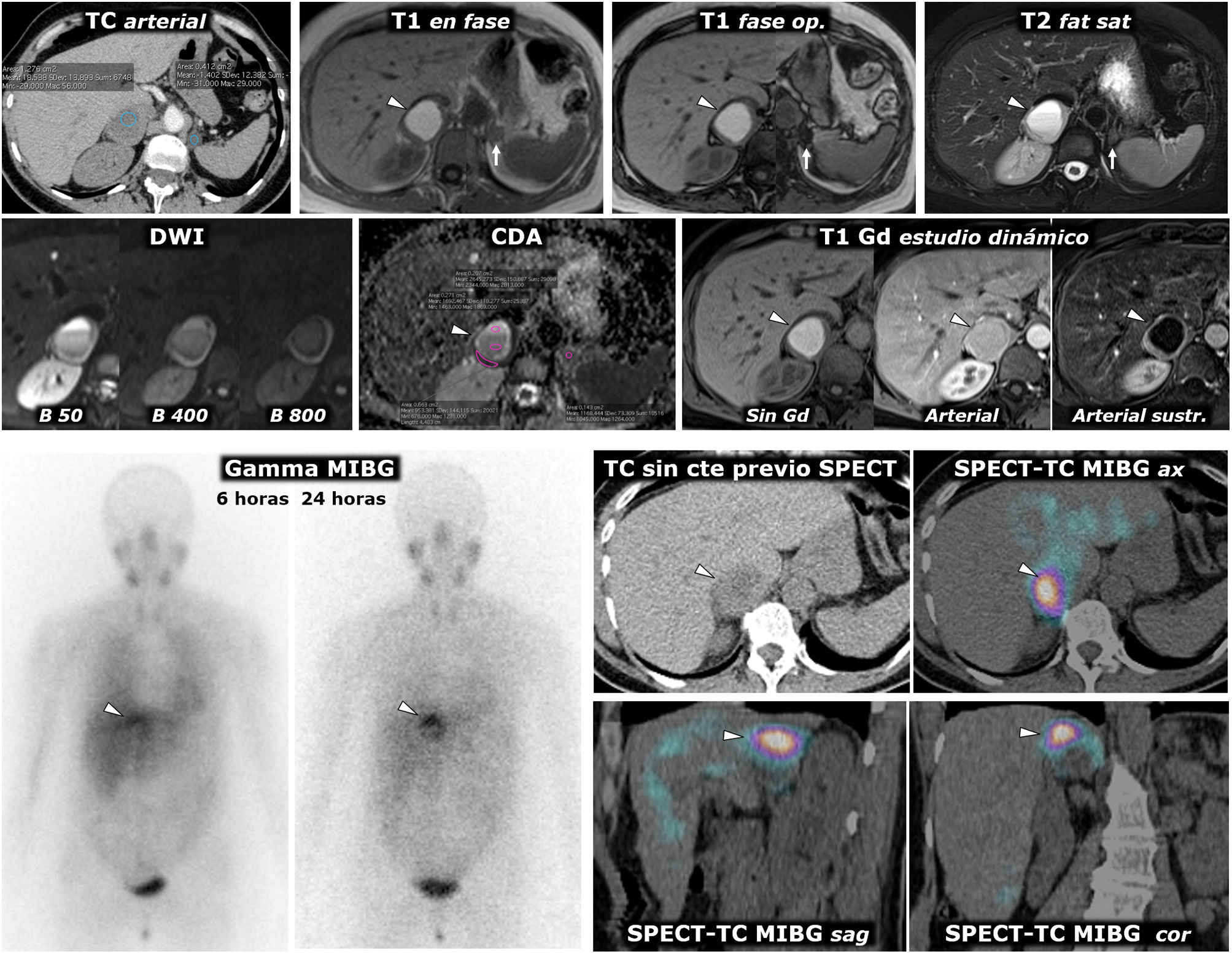
, uno mayor derecho, con valores de atenuación medios algo inferiores a 20 UH, inespecífico, y otro menor izquierdo de –1,4 UH, que se caracteriza como adenoma rico en lípidos. En la RM se comprueba que la lesión derecha (cabezas de flecha) consiste en una lesión casi puramente hemorrágica, con alta señal en T1, sin caída de señal en fase opuesta en el interior ni en su periferia. El adenoma adrenal izquierdo (flechas) sí pierde intensamente señal en fase opuesta (se ha realizado un montaje de imágenes, para mostrar el centro de las 2 lesiones en la misma imagen). La lesión adrenal derecha presenta alta señal en T2, con 2 niveles líquidos que también se manifiestan en la secuencia potenciada en difusión y en el mapa de CDA, con restricción de la difusión progresivamente mayor hacia áreas gravitacionales. En la pared de esta lesión los valores medios de CDA son de 0,95 x 10–3 mm2/s. El adenoma presenta valores medios de 1,17. Como sucede en otras lesiones hemorrágicas, el reconocimiento de los realces parietales o nodulares se facilita con la sustracción de la imagen basal. Parte inferior: hallazgos en gammagrafía y SPET-TC con (123I) MIBG. En el estudio gammagráfico se aprecia captación por parte de la lesión derecha (cabezas de flecha), de forma más evidente en el estudio a las 24h de la administración del radiofármaco. El estudio SPECT-TC con el mismo radiofármaco, adquirido a las 6h, muestra de forma más clara la captación por parte de la lesión, que prácticamente confirma el diagnóstico de feocromocitoma. El adenoma izquierdo no capta.") Figura 3.
Figura 3.Feocromocitoma como masa casi puramente hemorrágica demostrado por Medicina Nuclear. Mujer de 58 años. Acude a Urgencias por cefalea y palpitaciones. Parte superior: TC y RM. Sendos nódulos adrenales (ROI elípticas en TC), uno mayor derecho, con valores de atenuación medios algo inferiores a 20 UH, inespecífico, y otro menor izquierdo de –1,4 UH, que se caracteriza como adenoma rico en lípidos. En la RM se comprueba que la lesión derecha (cabezas de flecha) consiste en una lesión casi puramente hemorrágica, con alta señal en T1, sin caída de señal en fase opuesta en el interior ni en su periferia. El adenoma adrenal izquierdo (flechas) sí pierde intensamente señal en fase opuesta (se ha realizado un montaje de imágenes, para mostrar el centro de las 2 lesiones en la misma imagen). La lesión adrenal derecha presenta alta señal en T2, con 2 niveles líquidos que también se manifiestan en la secuencia potenciada en difusión y en el mapa de CDA, con restricción de la difusión progresivamente mayor hacia áreas gravitacionales. En la pared de esta lesión los valores medios de CDA son de 0,95 x 10–3 mm2/s. El adenoma presenta valores medios de 1,17. Como sucede en otras lesiones hemorrágicas, el reconocimiento de los realces parietales o nodulares se facilita con la sustracción de la imagen basal. Parte inferior: hallazgos en gammagrafía y SPET-TC con (123I) MIBG. En el estudio gammagráfico se aprecia captación por parte de la lesión derecha (cabezas de flecha), de forma más evidente en el estudio a las 24h de la administración del radiofármaco. El estudio SPECT-TC con el mismo radiofármaco, adquirido a las 6h, muestra de forma más clara la captación por parte de la lesión, que prácticamente confirma el diagnóstico de feocromocitoma. El adenoma izquierdo no capta.
(0.41MB). - –
(111In) octeótrido SPECT, con más sensibilidad en la detección de metástasis.
- –
(18F) FDG PET-TC, muy sensible para detectar metástasis, con baja especificidad (fig. 4).
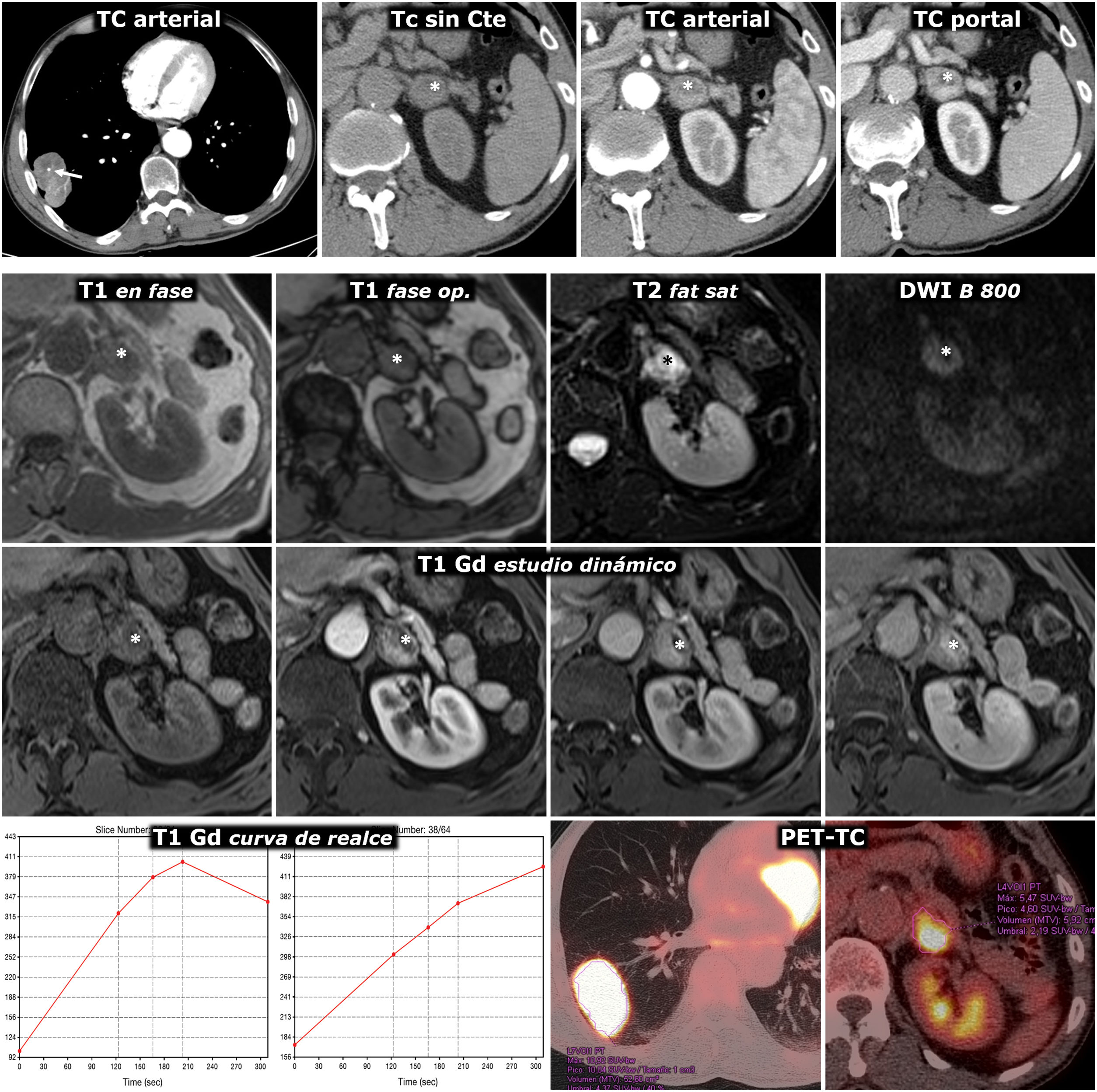
 y realce relativamente intenso y heterogéneo. Lesión nodular en la adrenal izquierda con realce también intenso, rápido y heterogéneo. En la fase basal del estudio presenta un área anterior con valores de atenuación inferiores a 10 UH (*). Aunque se interpreta que puede corresponder a un foco quístico o necrótico más que a contenido lipídico, pues esa región realza menos intensamente, se indica RM. Tres filas inferiores, RM y PET-TC: la región con menor densidad en TC se corresponde efectivamente con un foco quístico o necrótico (*): baja señal en T1, muy alta en T2, ausencia de restricción de la difusión del agua y ausencia de realce significativo. Las zonas sólidas de la lesión no pierden señal en fase opuesta, tienen alta señal en T2, muestran moderada restricción de la difusión y realzan intensamente, unas áreas de forma más rápida y otras más tardía, como se aprecia en las curvas de realce. El diagnóstico radiológico de presunción fue de metástasis. Se realizó (18F) FDG PET-TC, que mostró intensa actividad metabólica tanto en la lesión pulmonar como en la adrenal. El estudio histológico de la primera mostró un adenocarcinoma pulmonar y el de la segunda, un feocromocitoma. No hubo complicaciones quirúrgicas a pesar de que no se realizó preparación farmacológica.") Figura 4.
Figura 4.Error diagnóstico. Feocromocitoma adrenal interpretado como metástasis de adenocarcinoma de pulmón. Varón de 69 años, fumador. Hallazgo de masa pulmonar en la radiografía de tórax. Fila superior, TC: masa tumoral pulmonar lobulada y bien delimitada, con una calcificación puntiforme (flecha) y realce relativamente intenso y heterogéneo. Lesión nodular en la adrenal izquierda con realce también intenso, rápido y heterogéneo. En la fase basal del estudio presenta un área anterior con valores de atenuación inferiores a 10 UH (*). Aunque se interpreta que puede corresponder a un foco quístico o necrótico más que a contenido lipídico, pues esa región realza menos intensamente, se indica RM. Tres filas inferiores, RM y PET-TC: la región con menor densidad en TC se corresponde efectivamente con un foco quístico o necrótico (*): baja señal en T1, muy alta en T2, ausencia de restricción de la difusión del agua y ausencia de realce significativo. Las zonas sólidas de la lesión no pierden señal en fase opuesta, tienen alta señal en T2, muestran moderada restricción de la difusión y realzan intensamente, unas áreas de forma más rápida y otras más tardía, como se aprecia en las curvas de realce. El diagnóstico radiológico de presunción fue de metástasis. Se realizó (18F) FDG PET-TC, que mostró intensa actividad metabólica tanto en la lesión pulmonar como en la adrenal. El estudio histológico de la primera mostró un adenocarcinoma pulmonar y el de la segunda, un feocromocitoma. No hubo complicaciones quirúrgicas a pesar de que no se realizó preparación farmacológica.
(0.72MB). - –
(68Ga) DOTATATE PET-TC, ocasionalmente más sensible para detectar enfermedad metastásica, y con mayor resolución espacial.
, uno mayor derecho, con valores de atenuación medios algo inferiores a 20 UH, inespecífico, y otro menor izquierdo de –1,4 UH, que se caracteriza como adenoma rico en lípidos. En la RM se comprueba que la lesión derecha (cabezas de flecha) consiste en una lesión casi puramente hemorrágica, con alta señal en T1, sin caída de señal en fase opuesta en el interior ni en su periferia. El adenoma adrenal izquierdo (flechas) sí pierde intensamente señal en fase opuesta (se ha realizado un montaje de imágenes, para mostrar el centro de las 2 lesiones en la misma imagen). La lesión adrenal derecha presenta alta señal en T2, con 2 niveles líquidos que también se manifiestan en la secuencia potenciada en difusión y en el mapa de CDA, con restricción de la difusión progresivamente mayor hacia áreas gravitacionales. En la pared de esta lesión los valores medios de CDA son de 0,95 x 10–3 mm2/s. El adenoma presenta valores medios de 1,17. Como sucede en otras lesiones hemorrágicas, el reconocimiento de los realces parietales o nodulares se facilita con la sustracción de la imagen basal. Parte inferior: hallazgos en gammagrafía y SPET-TC con (123I) MIBG. En el estudio gammagráfico se aprecia captación por parte de la lesión derecha (cabezas de flecha), de forma más evidente en el estudio a las 24h de la administración del radiofármaco. El estudio SPECT-TC con el mismo radiofármaco, adquirido a las 6h, muestra de forma más clara la captación por parte de la lesión, que prácticamente confirma el diagnóstico de feocromocitoma. El adenoma izquierdo no capta.")
 y realce relativamente intenso y heterogéneo. Lesión nodular en la adrenal izquierda con realce también intenso, rápido y heterogéneo. En la fase basal del estudio presenta un área anterior con valores de atenuación inferiores a 10 UH (*). Aunque se interpreta que puede corresponder a un foco quístico o necrótico más que a contenido lipídico, pues esa región realza menos intensamente, se indica RM. Tres filas inferiores, RM y PET-TC: la región con menor densidad en TC se corresponde efectivamente con un foco quístico o necrótico (*): baja señal en T1, muy alta en T2, ausencia de restricción de la difusión del agua y ausencia de realce significativo. Las zonas sólidas de la lesión no pierden señal en fase opuesta, tienen alta señal en T2, muestran moderada restricción de la difusión y realzan intensamente, unas áreas de forma más rápida y otras más tardía, como se aprecia en las curvas de realce. El diagnóstico radiológico de presunción fue de metástasis. Se realizó (18F) FDG PET-TC, que mostró intensa actividad metabólica tanto en la lesión pulmonar como en la adrenal. El estudio histológico de la primera mostró un adenocarcinoma pulmonar y el de la segunda, un feocromocitoma. No hubo complicaciones quirúrgicas a pesar de que no se realizó preparación farmacológica.")
Actualmente no hay indicación para el muestreo hormonal selectivo en venas adrenales.
Hallazgos radiológicosPueden detectarse con ecografía (con diferentes ecogenicidades en su componente sólido), TC o RM. Las tablas 4 y 5 recogen, respectivamente, nuestros protocolos técnicos de TC y RM para el estudio de nódulos adrenales. Los hallazgos son inespecíficos, siendo orientativos estos datos20–26:
- –
Buena delimitación, incluso aunque se trate de tumores grandes (figs. 1-10). Un comportamiento infiltrativo se ha correlacionado con mayor agresividad biológica.
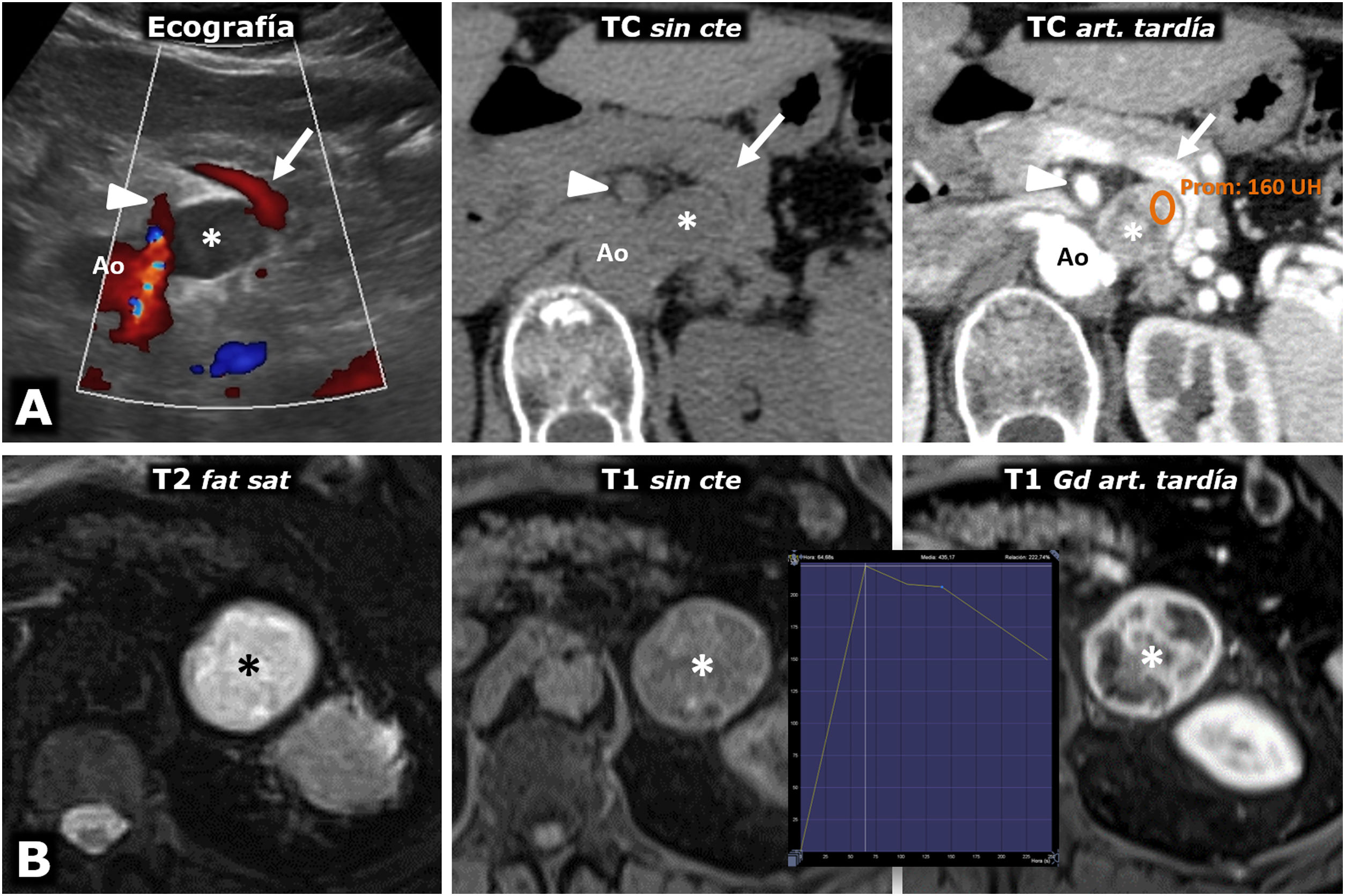
 Mujer de 74 años. Hallazgo incidental ecográfico de nódulo sólido hipoecogénico bien delimitado discretamente heterogéneo (*), situado entre la vena esplénica (flechas), la arteria mesentérica superior (cabezas de flecha) y la aorta (Ao). En la TC muestra realce intenso y heterogéneo en la fase arterial tardía, en la que alcanza las 160 UH en algunas áreas, con moderado lavado en fases posteriores (no mostrado). B) Varón de 73 años. En el curso de estudio por HTA grado II se encuentra una masa adrenal izquierda (*), que en la RM se comporta con alta intensidad de señal en T2, con marcado realce (relativo superior a 220%) con pico en la fase arterial tardía del estudio dinámico. La HTA se corrigió tras adrenalectomía laparoscópica.") Figura 5.
Figura 5.Intenso realce en feocromocitomas. A) Mujer de 74 años. Hallazgo incidental ecográfico de nódulo sólido hipoecogénico bien delimitado discretamente heterogéneo (*), situado entre la vena esplénica (flechas), la arteria mesentérica superior (cabezas de flecha) y la aorta (Ao). En la TC muestra realce intenso y heterogéneo en la fase arterial tardía, en la que alcanza las 160 UH en algunas áreas, con moderado lavado en fases posteriores (no mostrado). B) Varón de 73 años. En el curso de estudio por HTA grado II se encuentra una masa adrenal izquierda (*), que en la RM se comporta con alta intensidad de señal en T2, con marcado realce (relativo superior a 220%) con pico en la fase arterial tardía del estudio dinámico. La HTA se corrigió tras adrenalectomía laparoscópica.
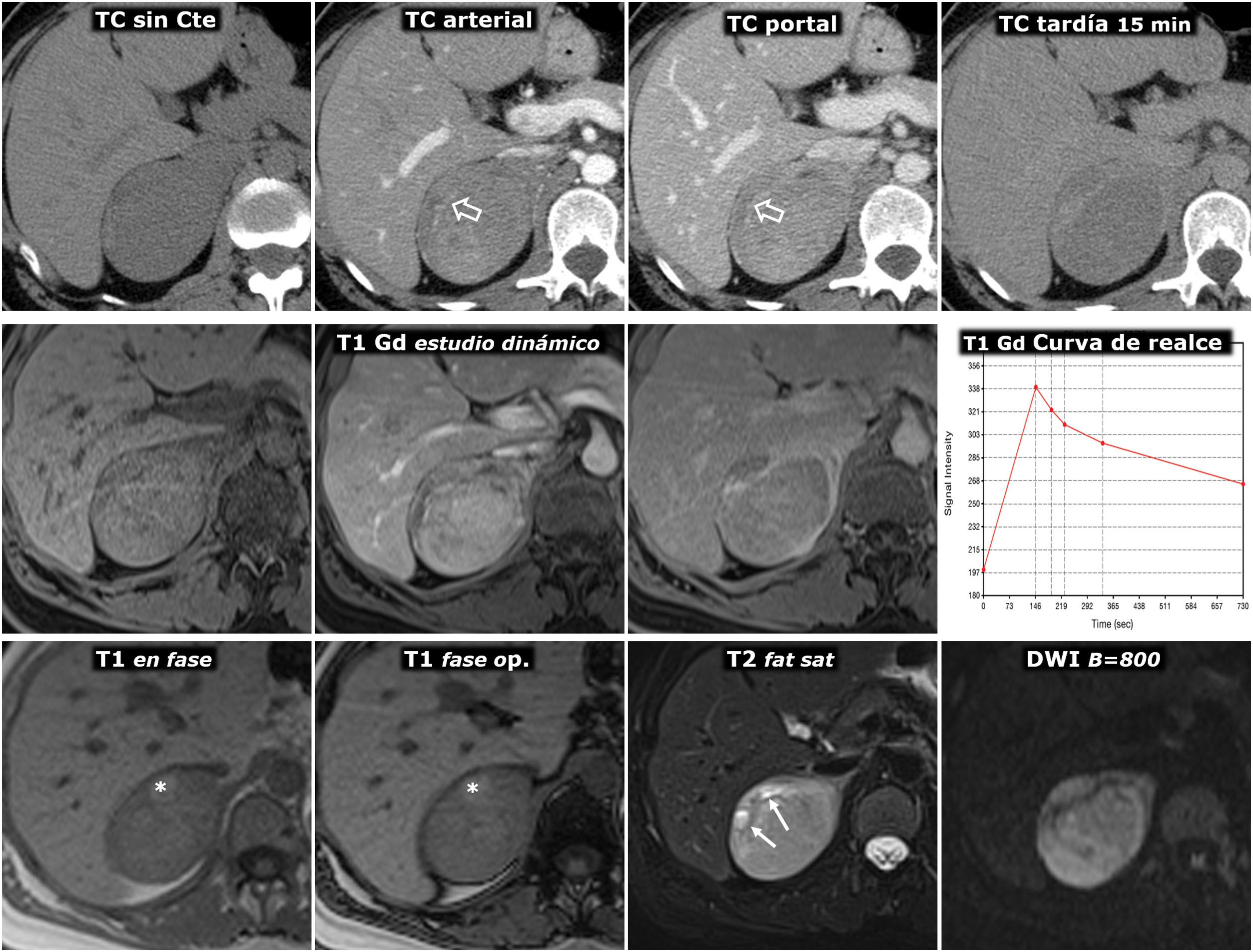
(0.48MB).. Los valores medios de atenuación en las distintas fases del estudio son de 30 (no demuestra presencia de lípidos), 63, 89 y 49 UH, respectivamente, con lavado absoluto del 67,8% y relativo del 44,9% en el estudio tardío, por encima, respectivamente, del 60 y el 40%, que sugieren adenoma con fiabilidad alta. Además, en algunos estudios se ha indicado que la presencia de vasos en el interior de una lesión nodular adrenal bien delimitada es muy indicativa de adenoma. No obstante, puesto que un tercio de los feocromocitomas también pueden presentar tasas de lavado similares a las de los adenomas, se consideró una lesión inespecífica y se realizó una RM. Fila central: estudio dinámico y curva de realce en RM con contraste extracelular. La masa muestra realce intenso (relativo superior a 330%), con pico en la fase arterial y lavado posterior. Fila inferior: otras secuencias de RM. No presenta caída de señal en fase opuesta, muestra alta señal en T2, que conserva en difusión con B alto y contiene algún pequeño foco quístico (flechas) y hemorrágico (*). La presencia de focos quísticos y hemorrágicos en una masa adrenal bien delimitada con el resto de características descritas llevó a un diagnóstico de presunción de feocromocitoma, que se apoyó en determinaciones elevadas de catecolaminas en orina y, posteriormente, se confirmó en el estudio histológico de la pieza quirúrgica.") Figura 6.
Figura 6.Feocromocitoma predominantemente sólido, con lavado tardío similar al adenoma en estudio de TC. Mujer de 51 años con hallazgo de una masa adrenal derecha en una ecografía por TEP. No presenta HTA. Fila superior, TC: masa adrenal derecha bien delimitada y bastante homogénea con un vaso en su interior (flechas huecas). Los valores medios de atenuación en las distintas fases del estudio son de 30 (no demuestra presencia de lípidos), 63, 89 y 49 UH, respectivamente, con lavado absoluto del 67,8% y relativo del 44,9% en el estudio tardío, por encima, respectivamente, del 60 y el 40%, que sugieren adenoma con fiabilidad alta. Además, en algunos estudios se ha indicado que la presencia de vasos en el interior de una lesión nodular adrenal bien delimitada es muy indicativa de adenoma. No obstante, puesto que un tercio de los feocromocitomas también pueden presentar tasas de lavado similares a las de los adenomas, se consideró una lesión inespecífica y se realizó una RM. Fila central: estudio dinámico y curva de realce en RM con contraste extracelular. La masa muestra realce intenso (relativo superior a 330%), con pico en la fase arterial y lavado posterior. Fila inferior: otras secuencias de RM. No presenta caída de señal en fase opuesta, muestra alta señal en T2, que conserva en difusión con B alto y contiene algún pequeño foco quístico (flechas) y hemorrágico (*). La presencia de focos quísticos y hemorrágicos en una masa adrenal bien delimitada con el resto de características descritas llevó a un diagnóstico de presunción de feocromocitoma, que se apoyó en determinaciones elevadas de catecolaminas en orina y, posteriormente, se confirmó en el estudio histológico de la pieza quirúrgica.
(0.49MB). y ecogenicidad heterogénea en las áreas sólidas, con zonas más ecogénicas y otras hipoecogénicas. Se sugiere feocromocitoma en el diagnóstico ecográfico. El aspecto en la RM es también característico, con áreas sólidas predominantes con alta señal en T2 (compárese con la de la musculatura paravertebral), que realzan intensamente y no muestran caída de señal en fase opuesta. Hay correlación entre las áreas más hipoecogénicas y las zonas con mayor restricción de la difusión del agua, que conservan mayor señal en la secuencia potenciada en difusión con B alto y son marcadamente hipointensas en el mapa paramétrico de CDA (con valores de 0,7 x 10–3 mm2/s). Las áreas quísticas son hipointensas en T1, hiperintensas en T2, no realzan y presentan una difusión del agua facilitada, con gran caída de señal con B alto y CDA de 2,2 x 10–3 mm2/s.") Figura 7.
Figura 7.Feocromocitoma con cambios quísticos y alta señal en T2. Ecografía y RM. Mujer de 30 años que precisa ingreso en la UCI por urgencia hipertensiva, con gran cefalea. En la ecografía se detecta una masa adrenal derecha prácticamente esférica, perfectamente delimitada, con pequeños focos quísticos (*) y ecogenicidad heterogénea en las áreas sólidas, con zonas más ecogénicas y otras hipoecogénicas. Se sugiere feocromocitoma en el diagnóstico ecográfico. El aspecto en la RM es también característico, con áreas sólidas predominantes con alta señal en T2 (compárese con la de la musculatura paravertebral), que realzan intensamente y no muestran caída de señal en fase opuesta. Hay correlación entre las áreas más hipoecogénicas y las zonas con mayor restricción de la difusión del agua, que conservan mayor señal en la secuencia potenciada en difusión con B alto y son marcadamente hipointensas en el mapa paramétrico de CDA (con valores de 0,7 x 10–3 mm2/s). Las áreas quísticas son hipointensas en T1, hiperintensas en T2, no realzan y presentan una difusión del agua facilitada, con gran caída de señal con B alto y CDA de 2,2 x 10–3 mm2/s.
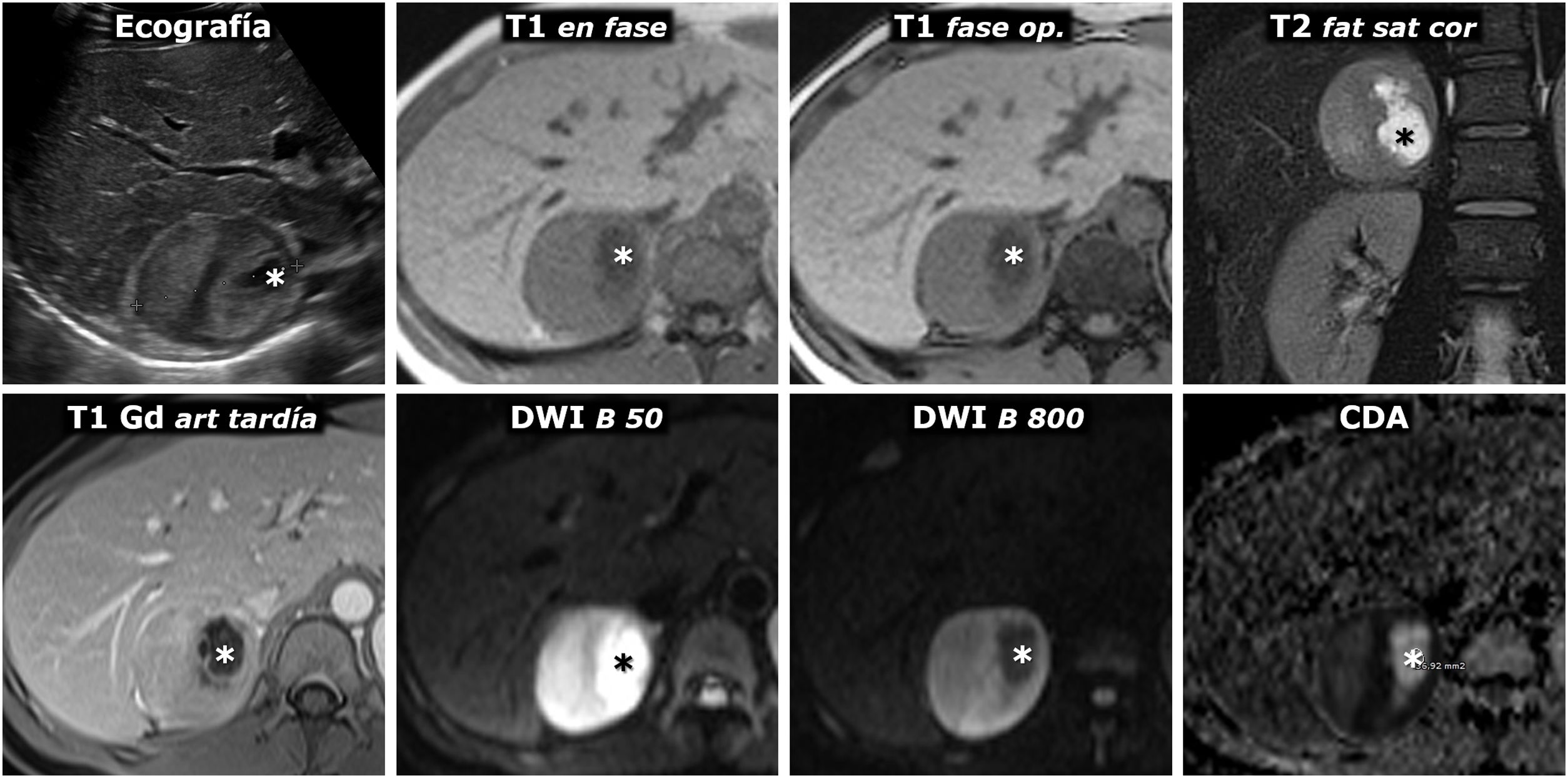
(0.27MB)., que en el mapa de CDA muestran alta señal por ausencia de restricción de la difusión del agua. Estos focos pierden por lo mismo la señal en la secuencia potenciada en difusión con valor alto de B. En el estudio dinámico con contraste la periferia de la lesión muestra realce intenso, con el pico en la fase arterial del estudio, mientras que el centro, con fenómenos quísticos que se demostraron anatomopatológicamente, tiene escaso realce.") Figura 8.
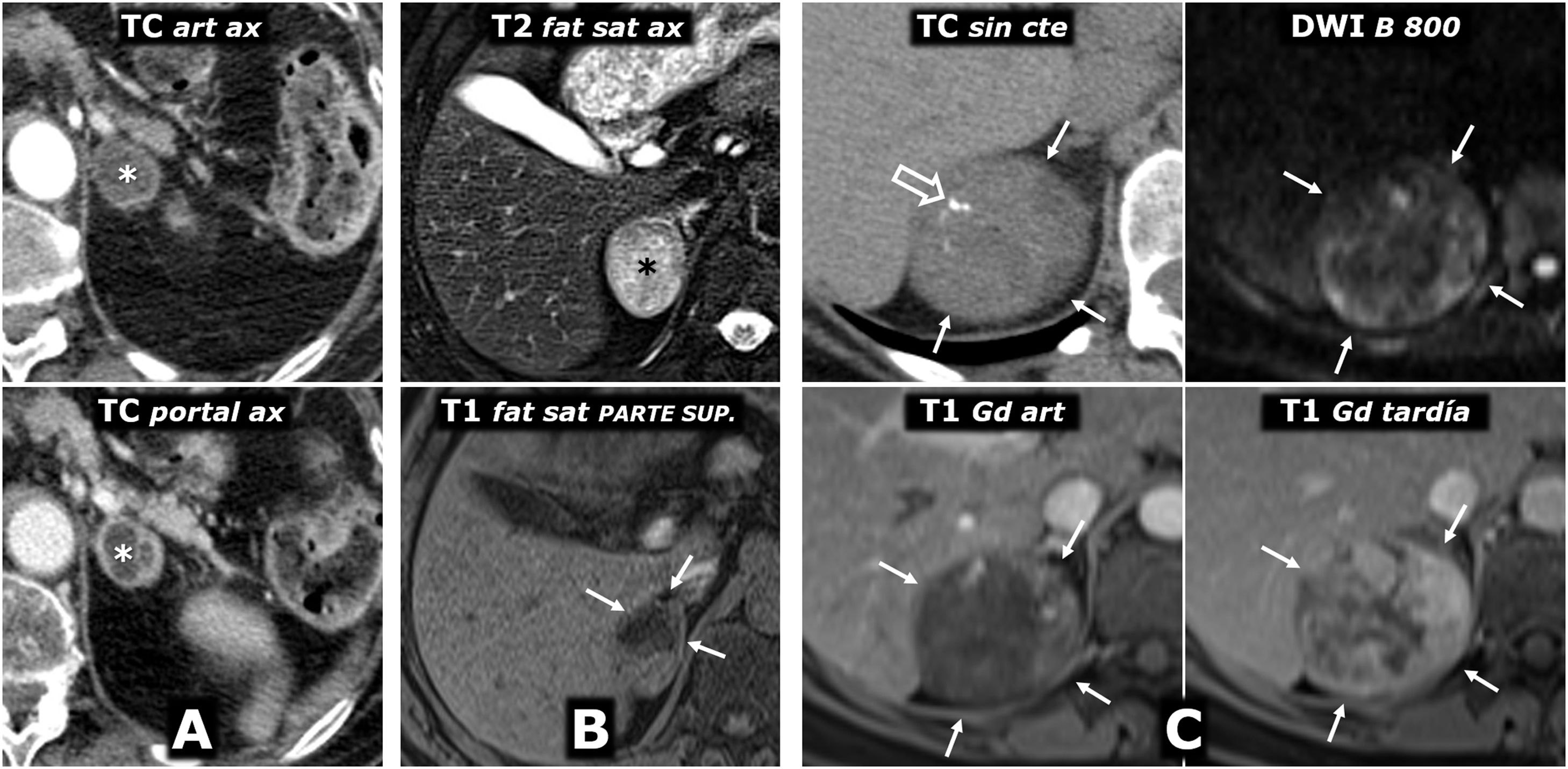
Figura 8.Hallazgo incidental de feocromocitoma adrenal con áreas quísticas en estudio por traumatismo abdominal. Varón de 57 años. En la TC en fase portal se identifica incidentalmente una lesión nodular adrenal derecha con un centro más hipoatenuante y una periferia hiperatenuante, probablemente por captación de contraste. En la RM no presenta caída de señal en fase opuesta. Es marcadamente hiperintensa en T2, con algunos focos de aspecto quístico (flechas), que en el mapa de CDA muestran alta señal por ausencia de restricción de la difusión del agua. Estos focos pierden por lo mismo la señal en la secuencia potenciada en difusión con valor alto de B. En el estudio dinámico con contraste la periferia de la lesión muestra realce intenso, con el pico en la fase arterial del estudio, mientras que el centro, con fenómenos quísticos que se demostraron anatomopatológicamente, tiene escaso realce.
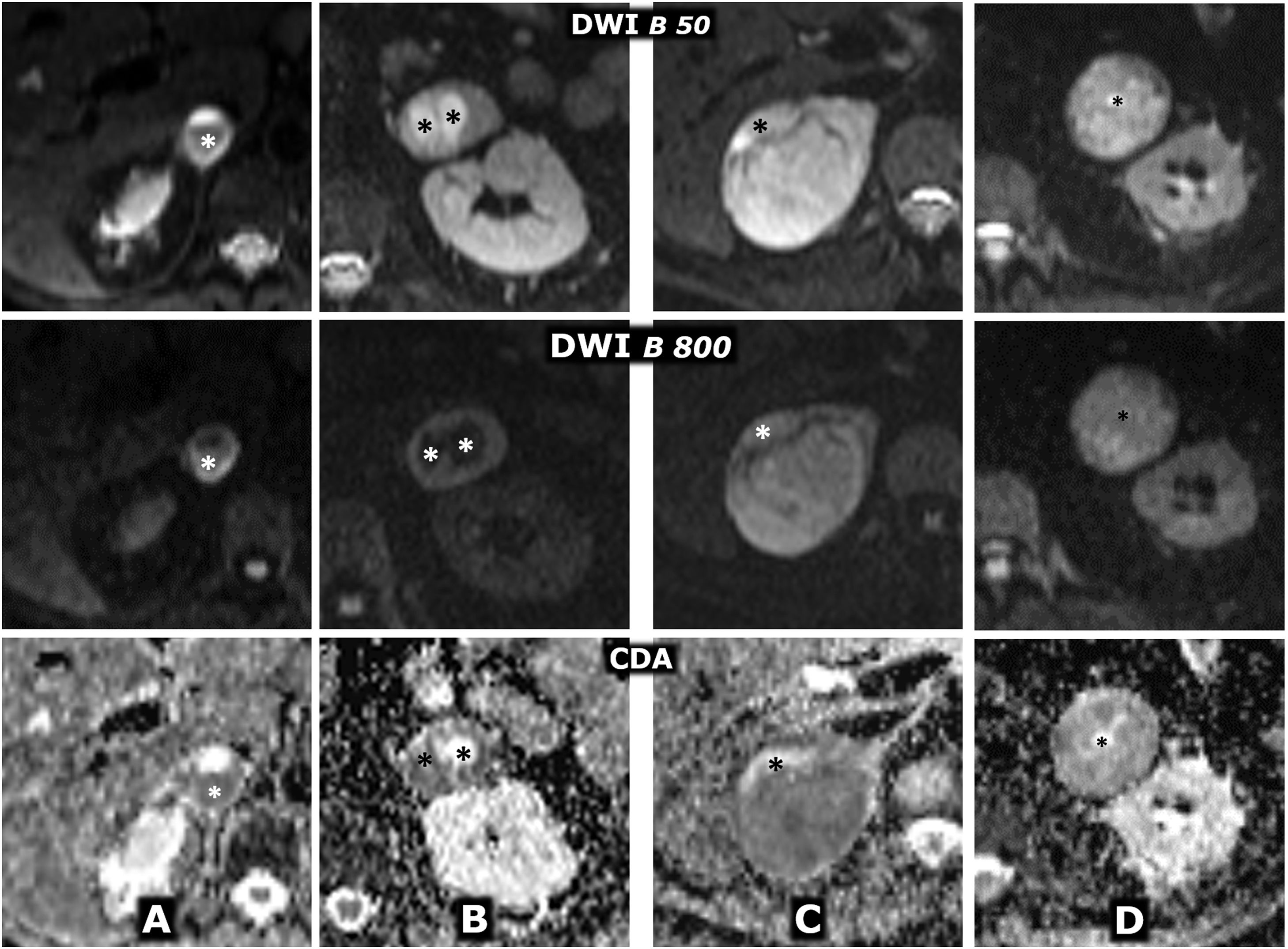
(0.29MB).. Fila central: secuencia de difusión con B=800 s/mm2 (potenciación en difusión). Fila inferior: mapa de coeficiente de difusión aparente (CDA). A) Mujer de 64 años con crisis hipertensivas y paroxismos de cefalea. Feocromocitoma adrenal derecho de 22mm con fenómenos quístico-hemorrágicos casi globales, demostrados histológicamente, con un área antigravitacional con difusión del agua facilitada y otra gravitacional (*) con baja señal en T2 y T1 (no mostrado), relativamente alta en difusión con B=800 s/mm2 y baja en el mapa de CDA, con valor promedio de 0,9 x 10–3 mm2/s. La pared de tejido sólido, de unos 3mm de grosor, muestra alta señal en T2 y restricción de la difusión, con alta señal con B=800 y CDA promedio de 0,83. Índice Ki 67 en la pieza quirúrgica: 2%. No ha presentado recidiva o progresión 20 meses después. B) Varón de 49 años con diagnóstico incidental de feocromocitoma izquierdo de 43mm, confirmado por elevación de catecolaminas en suero y orina e histológicamente tras resección laparoscópica. Masa predominantemente sólida, con 2 áreas quísticas (*) donde el agua presenta difusión libre. En áreas sólidas periféricas hay restricción de la difusión, con CDA promedio de 0,78 x 103 mm2/s. Índice Ki 67:<1%. Sin recidiva o progresión 7años después. C) Mujer de 51 años con crisis hipertensivas. Feocromocitoma derecho prácticamente sólido de 82mm, con pequeños focos quísticos anteriores (*) con la difusión del agua facilitada. En áreas sólidas hay muy alta señal en T2 y moderada restricción de la difusión del agua, con valor de CDA promedio de 0,94 x 10-3 mm2/s. Índice Ki 67: 3%. Sin recidiva o progresión 6 años después. D) Varón de 73 años con HTA grado II. Feocromocitoma izquierdo, prácticamente sólido, de 51mm, solo, con muy pequeños focos centrales de degeneración quística. Las áreas sólidas presentan señal alta en T2 y relativamente escasa restricción de la difusión del agua, con CDA de 1,3 x 10–3 mm2/s. Índice Ki 67: 2%. Sin recidiva o progresión un año tras la exéresis.") Figura 9.
Figura 9.Comportamiento variable en difusión en 4 casos diferentes de feocromocitoma. Fila superior: secuencia de difusión con B=50 s/mm2 (potenciación casi completamente en T2). Fila central: secuencia de difusión con B=800 s/mm2 (potenciación en difusión). Fila inferior: mapa de coeficiente de difusión aparente (CDA). A) Mujer de 64 años con crisis hipertensivas y paroxismos de cefalea. Feocromocitoma adrenal derecho de 22mm con fenómenos quístico-hemorrágicos casi globales, demostrados histológicamente, con un área antigravitacional con difusión del agua facilitada y otra gravitacional (*) con baja señal en T2 y T1 (no mostrado), relativamente alta en difusión con B=800 s/mm2 y baja en el mapa de CDA, con valor promedio de 0,9 x 10–3 mm2/s. La pared de tejido sólido, de unos 3mm de grosor, muestra alta señal en T2 y restricción de la difusión, con alta señal con B=800 y CDA promedio de 0,83. Índice Ki 67 en la pieza quirúrgica: 2%. No ha presentado recidiva o progresión 20 meses después. B) Varón de 49 años con diagnóstico incidental de feocromocitoma izquierdo de 43mm, confirmado por elevación de catecolaminas en suero y orina e histológicamente tras resección laparoscópica. Masa predominantemente sólida, con 2 áreas quísticas (*) donde el agua presenta difusión libre. En áreas sólidas periféricas hay restricción de la difusión, con CDA promedio de 0,78 x 103 mm2/s. Índice Ki 67:<1%. Sin recidiva o progresión 7años después. C) Mujer de 51 años con crisis hipertensivas. Feocromocitoma derecho prácticamente sólido de 82mm, con pequeños focos quísticos anteriores (*) con la difusión del agua facilitada. En áreas sólidas hay muy alta señal en T2 y moderada restricción de la difusión del agua, con valor de CDA promedio de 0,94 x 10-3 mm2/s. Índice Ki 67: 3%. Sin recidiva o progresión 6 años después. D) Varón de 73 años con HTA grado II. Feocromocitoma izquierdo, prácticamente sólido, de 51mm, solo, con muy pequeños focos centrales de degeneración quística. Las áreas sólidas presentan señal alta en T2 y relativamente escasa restricción de la difusión del agua, con CDA de 1,3 x 10–3 mm2/s. Índice Ki 67: 2%. Sin recidiva o progresión un año tras la exéresis.
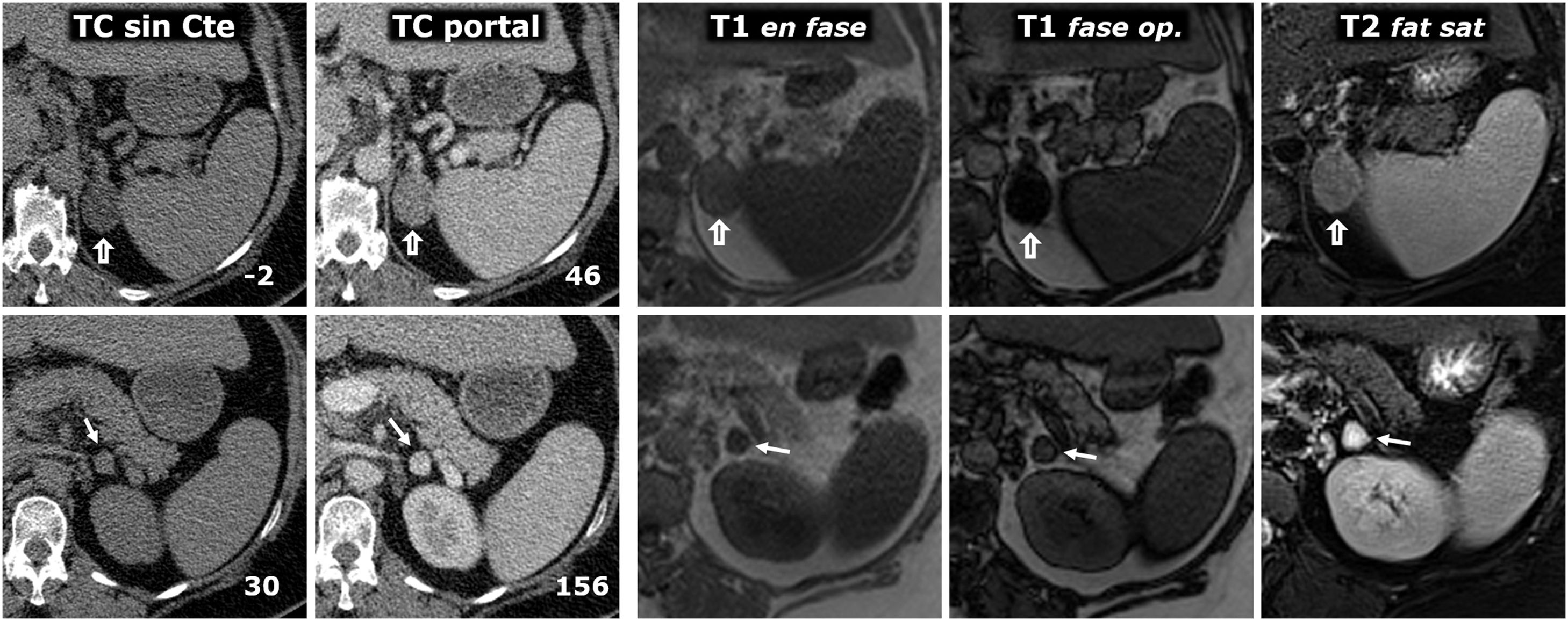
(0.5MB). y en la inferior, el feocromocitoma (flechas), ambos confirmados histológicamente. Dos columnas de la izquierda, TC: el número en la parte inferior derecha de cada recuadro corresponde al valor medio de atenuación de la lesión en el corte axial que se muestra. El adenoma presenta atenuación basal negativa, lo que permite su caracterización. La del feocromocitoma es de 30 UH, inespecífica. Realza intensamente en la fase arterial, de forma heterogénea (no mostrado), y en la portal, de forma más homogénea, rebasando las 130 UH. No se realizó estudio de lavado tardío. Resto de columnas a la derecha, RM: el adenoma presenta una gran caída de señal en fase opuesta, que delata su contenido en lípidos microscópicos. El feocromocitoma no muestra una caída de señal significativa y se rodea de artefacto «en tinta china». El estudio cuantitativo tampoco muestra un contenido lipídico significativo. El adenoma tiene una señal intermedia en T2. El feocromocitoma muestra una señal muy alta («signo de la bombilla»).") Figura 10.
Figura 10.Tumor de colisión: adenoma y feocromocitoma en la misma glándula adrenal. Varón de 58 años. Hallazgo incidental de 2 nódulos en la adrenal izquierda. En la fila superior se estudia el adenoma (flechas huecas) y en la inferior, el feocromocitoma (flechas), ambos confirmados histológicamente. Dos columnas de la izquierda, TC: el número en la parte inferior derecha de cada recuadro corresponde al valor medio de atenuación de la lesión en el corte axial que se muestra. El adenoma presenta atenuación basal negativa, lo que permite su caracterización. La del feocromocitoma es de 30 UH, inespecífica. Realza intensamente en la fase arterial, de forma heterogénea (no mostrado), y en la portal, de forma más homogénea, rebasando las 130 UH. No se realizó estudio de lavado tardío. Resto de columnas a la derecha, RM: el adenoma presenta una gran caída de señal en fase opuesta, que delata su contenido en lípidos microscópicos. El feocromocitoma no muestra una caída de señal significativa y se rodea de artefacto «en tinta china». El estudio cuantitativo tampoco muestra un contenido lipídico significativo. El adenoma tiene una señal intermedia en T2. El feocromocitoma muestra una señal muy alta («signo de la bombilla»).
(0.42MB). - –
Realce intenso y rápido con contraste yodado (suelen alcanzar más de 130 UH); o paramagnético, no contraindicados (fig. 5). Puede solaparse con el del adenoma.
- –
Lavado tardío menor que el del adenoma, aunque en el 35% de los casos los valores se solapan25,26 (fig. 6). En el 65% restante no permite diferenciarlo del carcinoma y las metástasis.
- –
Alta señal en secuencia T2 («signo de la bombilla») en áreas tumorales sólidas, con alta especificidad, pero baja sensibilidad (figs. 5 y 7). La medición de la ratio de señal en secuencia T2 respecto a la musculatura paravertebral podría ser más útil que la valoración cualitativa25–27.
- –
Frecuentes cambios quísticos y hemorrágicos (figs. 1-3, 7 y 8), lo que los hace heterogéneos, sobre todo cuando son grandes. El contenido puede ser hiperintenso en secuencia T1 por hemorragia (fig. 3). Pueden llegar a tener un aspecto puramente quístico o mostrar solo un anillo periférico de tumor sólido, con un realce y comportamiento en secuencia T2 conforme a lo descrito. En ocasiones la valoración del realce puede requerir sustracción de la imagen basal o cuantificación espectral.
- –
Ausencia de lípidos macro o microscópicos. Las áreas sólidas tienen en TC sin contraste valores de atenuación mayores de 10 UH (figs. 2, 4 y 6), pero pueden ser menores en regiones quísticas. La presencia de focos macroscópicos de grasa en un feocromocitoma es excepcional (menos del 0,5% de los casos). La ausencia de caída de señal significativa en fase opuesta (figs. 3, 4, 6, 7 y 8) es uno de los datos que permiten un diagnóstico diferencial más seguro con el adenoma25. Las áreas con alta señal en secuencia T1 en RM, frecuentes por fenómenos hemorrágicos, no se suprimen con saturación grasa.
- –
El comportamiento en secuencia potenciada en difusión no aporta un gran valor diagnóstico. Las áreas quísticas o necróticas muestran generalmente difusión libre, con valores altos de coeficiente de difusión aparente (CDA), y las áreas sólidas suelen tener la difusión restringida en mayor o menor grado (fig. 9). En general, existe solapamiento en los valores de CDA del feocromocitoma y otros tumores adrenales, incluyendo los adenomas. La excepción son los carcinomas y los linfomas adrenales primarios, que presentan una marcada restricción de la difusión. Algunos estudios han documentado valores más altos de CDA que en adenomas o metástasis, diferencias de CDA entre benignos y malignos o alteraciones del histograma, pero dichos hallazgos no están confirmados.
- –
Pueden presentar calcificaciones (hasta un 10% en la literatura, aunque en nuestra experiencia muy infrecuentes), en general puntiformes.
- –
Es excepcional la coexistencia de un feocromocitoma y otro tumor en la misma adrenal (tumor de colisión), el menos infrecuente el adenoma (fig. 10).
 Mujer de 74 años. Hallazgo incidental ecográfico de nódulo sólido hipoecogénico bien delimitado discretamente heterogéneo (*), situado entre la vena esplénica (flechas), la arteria mesentérica superior (cabezas de flecha) y la aorta (Ao). En la TC muestra realce intenso y heterogéneo en la fase arterial tardía, en la que alcanza las 160 UH en algunas áreas, con moderado lavado en fases posteriores (no mostrado). B) Varón de 73 años. En el curso de estudio por HTA grado II se encuentra una masa adrenal izquierda (*), que en la RM se comporta con alta intensidad de señal en T2, con marcado realce (relativo superior a 220%) con pico en la fase arterial tardía del estudio dinámico. La HTA se corrigió tras adrenalectomía laparoscópica.")
. Los valores medios de atenuación en las distintas fases del estudio son de 30 (no demuestra presencia de lípidos), 63, 89 y 49 UH, respectivamente, con lavado absoluto del 67,8% y relativo del 44,9% en el estudio tardío, por encima, respectivamente, del 60 y el 40%, que sugieren adenoma con fiabilidad alta. Además, en algunos estudios se ha indicado que la presencia de vasos en el interior de una lesión nodular adrenal bien delimitada es muy indicativa de adenoma. No obstante, puesto que un tercio de los feocromocitomas también pueden presentar tasas de lavado similares a las de los adenomas, se consideró una lesión inespecífica y se realizó una RM. Fila central: estudio dinámico y curva de realce en RM con contraste extracelular. La masa muestra realce intenso (relativo superior a 330%), con pico en la fase arterial y lavado posterior. Fila inferior: otras secuencias de RM. No presenta caída de señal en fase opuesta, muestra alta señal en T2, que conserva en difusión con B alto y contiene algún pequeño foco quístico (flechas) y hemorrágico (*). La presencia de focos quísticos y hemorrágicos en una masa adrenal bien delimitada con el resto de características descritas llevó a un diagnóstico de presunción de feocromocitoma, que se apoyó en determinaciones elevadas de catecolaminas en orina y, posteriormente, se confirmó en el estudio histológico de la pieza quirúrgica.")
 y ecogenicidad heterogénea en las áreas sólidas, con zonas más ecogénicas y otras hipoecogénicas. Se sugiere feocromocitoma en el diagnóstico ecográfico. El aspecto en la RM es también característico, con áreas sólidas predominantes con alta señal en T2 (compárese con la de la musculatura paravertebral), que realzan intensamente y no muestran caída de señal en fase opuesta. Hay correlación entre las áreas más hipoecogénicas y las zonas con mayor restricción de la difusión del agua, que conservan mayor señal en la secuencia potenciada en difusión con B alto y son marcadamente hipointensas en el mapa paramétrico de CDA (con valores de 0,7 x 10–3 mm2/s). Las áreas quísticas son hipointensas en T1, hiperintensas en T2, no realzan y presentan una difusión del agua facilitada, con gran caída de señal con B alto y CDA de 2,2 x 10–3 mm2/s.")
, que en el mapa de CDA muestran alta señal por ausencia de restricción de la difusión del agua. Estos focos pierden por lo mismo la señal en la secuencia potenciada en difusión con valor alto de B. En el estudio dinámico con contraste la periferia de la lesión muestra realce intenso, con el pico en la fase arterial del estudio, mientras que el centro, con fenómenos quísticos que se demostraron anatomopatológicamente, tiene escaso realce.")
. Fila central: secuencia de difusión con B=800 s/mm2 (potenciación en difusión). Fila inferior: mapa de coeficiente de difusión aparente (CDA). A) Mujer de 64 años con crisis hipertensivas y paroxismos de cefalea. Feocromocitoma adrenal derecho de 22mm con fenómenos quístico-hemorrágicos casi globales, demostrados histológicamente, con un área antigravitacional con difusión del agua facilitada y otra gravitacional (*) con baja señal en T2 y T1 (no mostrado), relativamente alta en difusión con B=800 s/mm2 y baja en el mapa de CDA, con valor promedio de 0,9 x 10–3 mm2/s. La pared de tejido sólido, de unos 3mm de grosor, muestra alta señal en T2 y restricción de la difusión, con alta señal con B=800 y CDA promedio de 0,83. Índice Ki 67 en la pieza quirúrgica: 2%. No ha presentado recidiva o progresión 20 meses después. B) Varón de 49 años con diagnóstico incidental de feocromocitoma izquierdo de 43mm, confirmado por elevación de catecolaminas en suero y orina e histológicamente tras resección laparoscópica. Masa predominantemente sólida, con 2 áreas quísticas (*) donde el agua presenta difusión libre. En áreas sólidas periféricas hay restricción de la difusión, con CDA promedio de 0,78 x 103 mm2/s. Índice Ki 67:<1%. Sin recidiva o progresión 7años después. C) Mujer de 51 años con crisis hipertensivas. Feocromocitoma derecho prácticamente sólido de 82mm, con pequeños focos quísticos anteriores (*) con la difusión del agua facilitada. En áreas sólidas hay muy alta señal en T2 y moderada restricción de la difusión del agua, con valor de CDA promedio de 0,94 x 10-3 mm2/s. Índice Ki 67: 3%. Sin recidiva o progresión 6 años después. D) Varón de 73 años con HTA grado II. Feocromocitoma izquierdo, prácticamente sólido, de 51mm, solo, con muy pequeños focos centrales de degeneración quística. Las áreas sólidas presentan señal alta en T2 y relativamente escasa restricción de la difusión del agua, con CDA de 1,3 x 10–3 mm2/s. Índice Ki 67: 2%. Sin recidiva o progresión un año tras la exéresis.")
 y en la inferior, el feocromocitoma (flechas), ambos confirmados histológicamente. Dos columnas de la izquierda, TC: el número en la parte inferior derecha de cada recuadro corresponde al valor medio de atenuación de la lesión en el corte axial que se muestra. El adenoma presenta atenuación basal negativa, lo que permite su caracterización. La del feocromocitoma es de 30 UH, inespecífica. Realza intensamente en la fase arterial, de forma heterogénea (no mostrado), y en la portal, de forma más homogénea, rebasando las 130 UH. No se realizó estudio de lavado tardío. Resto de columnas a la derecha, RM: el adenoma presenta una gran caída de señal en fase opuesta, que delata su contenido en lípidos microscópicos. El feocromocitoma no muestra una caída de señal significativa y se rodea de artefacto «en tinta china». El estudio cuantitativo tampoco muestra un contenido lipídico significativo. El adenoma tiene una señal intermedia en T2. El feocromocitoma muestra una señal muy alta («signo de la bombilla»).")
Protocolo técnico de TC para el estudio de nódulo adrenal
| Colimación | Factor Pitch | Grosor reconstrucción | Tiempo de rotación | Kilovoltaje | Miliamperaje | Contraste | Fases de estudio vascular |
|---|---|---|---|---|---|---|---|
| 0,6 mm | 0,6 | 1 mm | 0,5 s | 120 (100 u 80 en casos seleccionados) | 158 basales, modulación automática | Iobitridol 350 mgI/mlTasa: 3,5 cc/sCantidad: 1,8 cc/kg o hasta 140 ccDetección bolo en aorta abdominal | Sin contrasteaArterial tardía. Detección + 20 sPortal. Detección + 50 sTardía. Detección + 15 min |
Equipo: Somatom Definition AS+128®, Siemens Healthineers (Erlangen, Alemania).
Secuencias básicas de nuestro protocolo de estudio por RM para nódulos adrenales (algunos parámetros se ajustan a las condiciones del paciente)
| T1 en fase/fase opuesta | T2 | Difusión | T1 dinámico con contraste | |
|---|---|---|---|---|
| Tipo secuencia | GE | TSE | EPI | VIBE |
| Orientación | Axial | Axial | Axial | Axial |
| Grosor corte (mm) | 3-5 | 2-5 | 5 | 3 |
| TR (ms) | 163 | 1.600 | 1.500 | 4,9 |
| TE (ms) | 2,4 / 4,8 | 75 | 79 | 2,39 |
| FOV de lectura (mm) | 400 | 400 | 380 | 440 |
| FOV de fase (mm) | 75 | 75 | 75 | 72 |
| Detalles técnicos | Adquisición doble eco | Opcional: fatsat o STIROpcional: coronal | B 50, 400 y 800mm2/sSupresión grasa SPAIRMapa CDA | Gadobutrol 1 mmol Gd/ml0,1 ml/kg a 2-2,5 ml/minFases: 0 s-inyección-1min 06 s-1min 49 s-2min 26 s-4min 12 sPuede obviarse si adenoma en T1 doble eco |
| Respiración | Apnea | Navegación | Navegación | Apneas sucesivas |
| Tiempo aprox. | 18-30 s | 3min 29 s | 3min 38 s | 5 x 11-18 s (hasta 3min 30 s) |
Magnetom Avanto® 1,5 T, Siemens Healthineers (Erlangen, Alemania). El tiempo total de exploración del estudio abreviado está en torno a los 10min.
CDA: coeficiente de difusión aparente; fatsat: saturación espectral de la grasa; FOV: field of view (campo de visión); GE: gradient echo (eco de gradiente); EPI: echo-planar imaging (imagen eco planar); SPAIR: SPectral Attenuated Inversion Recovery (inversión-recuperación con atenuación espectral); STIR: short inversión time inversión recovery (inversión-recuperación con tiempo corto de inversión); TE: tiempo de eco; TR: tiempo de repetición; TSE: turbo spin-echo; VIBE: volume interpolated breath-hold examination (estudio en apnea con interpolación volumétrica).
Generalmente es imposible predecir agresividad histológica.
Recientemente se ha analizado en varios trabajos publicados28,29 (alguno multicéntrico29) la utilidad de la radiómica en estudios RM para diferenciar los feocromocitomas de otros tumores adrenales. La aplicación del análisis radiómico ha sido estudiada en el trabajo de Kong et al.29 mediante nomogramas incluyendo datos clínico-analíticos con resultados prometedores. Sin embargo, requiere una más amplia valoración prospectiva de los resultados.
Los PGEA presentan hallazgos similares a los descritos, en localizaciones diferentes (fig. 11).
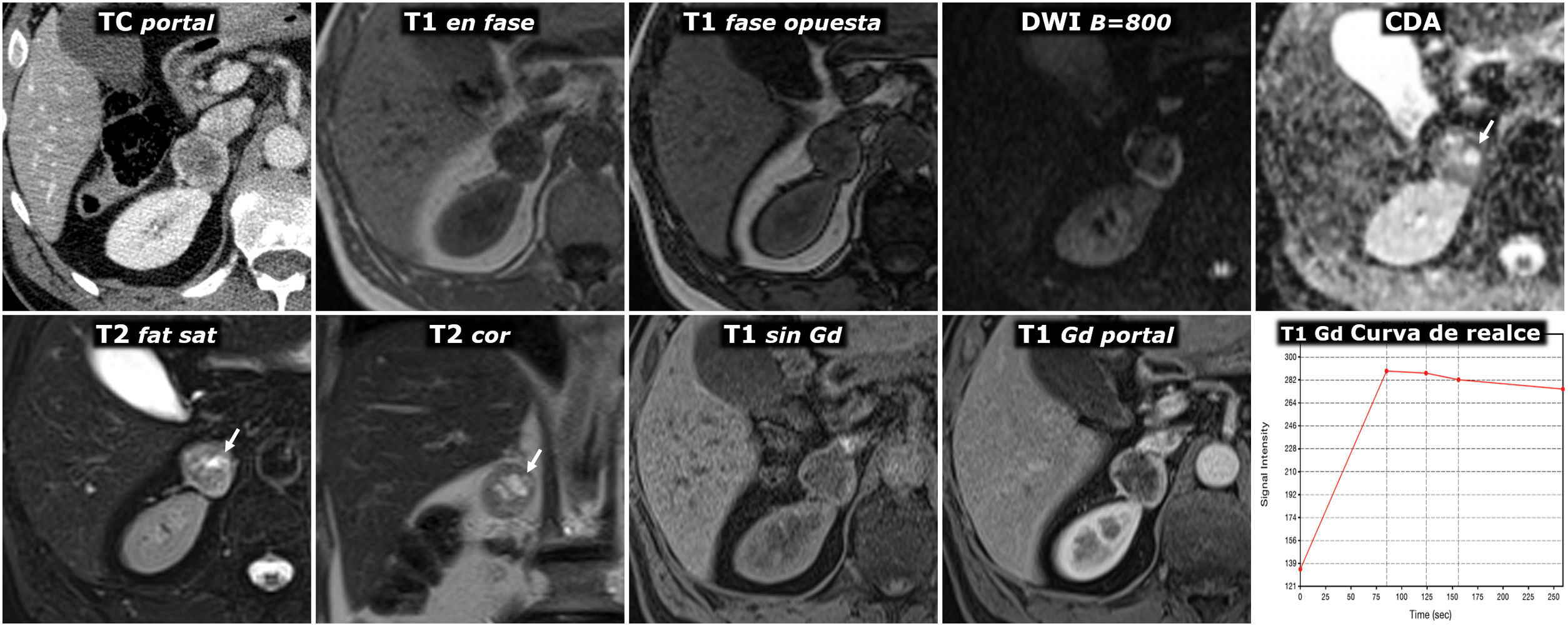
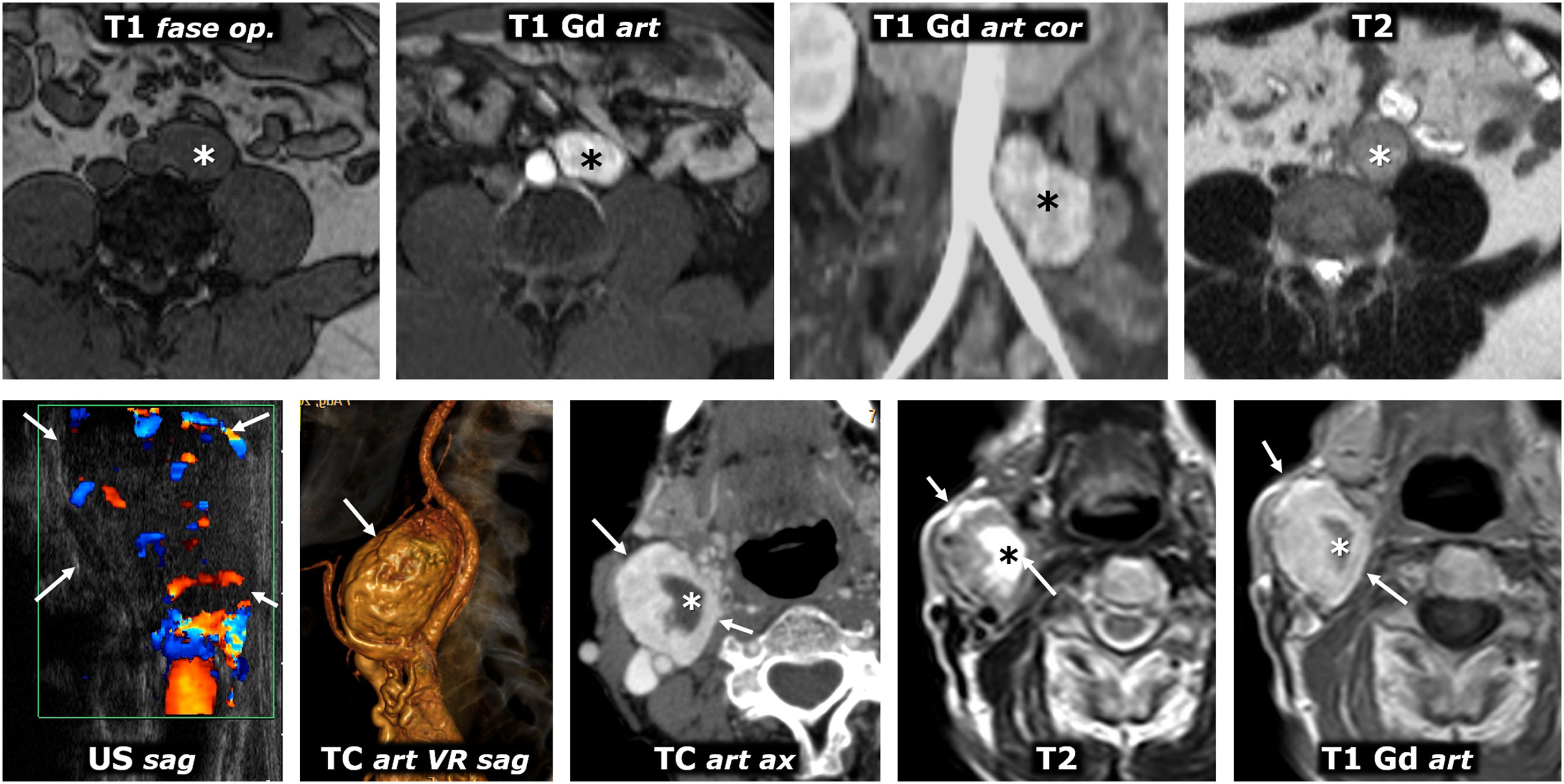
 situada a la izquierda de la aorta distal, inmediatamente por debajo del origen de la arteria mesentérica inferior, en la localización anatómica del órgano de Zuckerkandl. Esta lesión isointensa en T1 conserva su señal en fase opuesta, es hiperintensa en T2 y realza muy intensamente en la fase arterial del estudio con Gadolinio. Fila inferior: paraganglioma parasimpático no secretor del glomus carotídeo. Varón de 46 años. Consulta por masa cervical palpable. Lesión nodular (flechas) isoecogénica intensamente vascularizada junto a la bifurcación carotídea derecha (la imagen ecográfica se ha girado 90° para una mejor correlación). En TC presenta un realce intenso, con algunos focos quísticos en su interior (*). En la RM las áreas sólidas muestran alta señal en T2 e intermedia en T1, con realce intenso y rápido. Las regiones quísticas se comportan como el agua. Las determinaciones de catecolaminas fueron normales.")
Paragangliomas extraadrenales. Fila superior: paraganglioma simpático productor de metanefrinas del órgano de Zuckerkandl. Varón de 31 años en estudio por HTA y cefalea. Lesión nodular lobulada (*) situada a la izquierda de la aorta distal, inmediatamente por debajo del origen de la arteria mesentérica inferior, en la localización anatómica del órgano de Zuckerkandl. Esta lesión isointensa en T1 conserva su señal en fase opuesta, es hiperintensa en T2 y realza muy intensamente en la fase arterial del estudio con Gadolinio. Fila inferior: paraganglioma parasimpático no secretor del glomus carotídeo. Varón de 46 años. Consulta por masa cervical palpable. Lesión nodular (flechas) isoecogénica intensamente vascularizada junto a la bifurcación carotídea derecha (la imagen ecográfica se ha girado 90° para una mejor correlación). En TC presenta un realce intenso, con algunos focos quísticos en su interior (*). En la RM las áreas sólidas muestran alta señal en T2 e intermedia en T1, con realce intenso y rápido. Las regiones quísticas se comportan como el agua. Las determinaciones de catecolaminas fueron normales.
Diagnóstico diferencial por imagen del feocromocitoma. Al margen de estos aspectos es imprescindible considerar el contexto clínico y analítico
| Lesión | Posibles factores de confusión | Factores diferenciales | |
|---|---|---|---|
| Tumor sólido maligno | Metástasis | Posibles fenómenos quístico-necróticosSolapamiento realce en metástasis de tumores hipervascularesFc no agresivos consumen glucosaDifusión inespecífica en general | Neoplasia diseminadaMultiplicidadRealce menor y más tardío en tumores no hipervascularesMenor intensidad de señal en T2 en áreas sólidas |
| Carcinoma | Fc agresivos pueden ser infiltrantes | Masa grande, lobulada, heterogénea, infiltranteRestricción muy marcada de la difusión en zonas sólidasRealce menor y más tardíoMenor intensidad de señal en T2 en áreas sólidas | |
| Linfoma | Frecuentes fenómenos quístico-hemorrágicos | En general lobulado y peor definidoRestricción muy marcada de la difusión en zonas sólidasRealce menor y más tardío | |
| Sarcoma | Frecuentes fenómenos quístico-necróticos | Masa grande, lobulada, heterogénea, infiltrante | |
| Tumor sólido benigno | Adenoma | Posibles fenómenos quístico-hemorrágicos (infrecuente)35% de Fc con lavado tardío similar en TCCambios quísticos en Fc con atenuación basal <10 UH | Caída de señal en fase opuesta en rango característicoRealce menor y más tardíoMenor intensidad de señal en T2 en áreas sólidas |
| Mielolipoma | Lesiones redondeadas u ovales bien delimitadas. Pequeño porcentaje pobres en grasa | Casi siempre grasa macroscópica en TC o RM en algún áreaExcepcional que tengan fenómenos quístico-necróticos | |
| Hemangioma | Lesiones redondeadas bien delimitadasAlta señal en T2 | Patrón de realce característico, similar al hepático | |
| Lesión quística benigna | Quiste | Áreas quísticas por definición | Componente sólido ausente o casi |
| Hematoma-seudoquiste | Áreas quísticas, a veces pared gruesa con calcificaciones | Antecedente clínicoComponente sólido escaso, hipo o avascular | |
| Quiste hidatídico | Presentación variable, con áreas quísticas si parásitos viables | Coexistencia frecuente de hidatidosis hepáticaVesículas hijas o membranas son muy características | |
| Tuberculosis | Áreas quístico-necróticas frecuentes, con realce periférico | Afectación bilateralAfectación de otros órganosAusencia de componente tumoral sólido |
Fc: feocromocitomas.
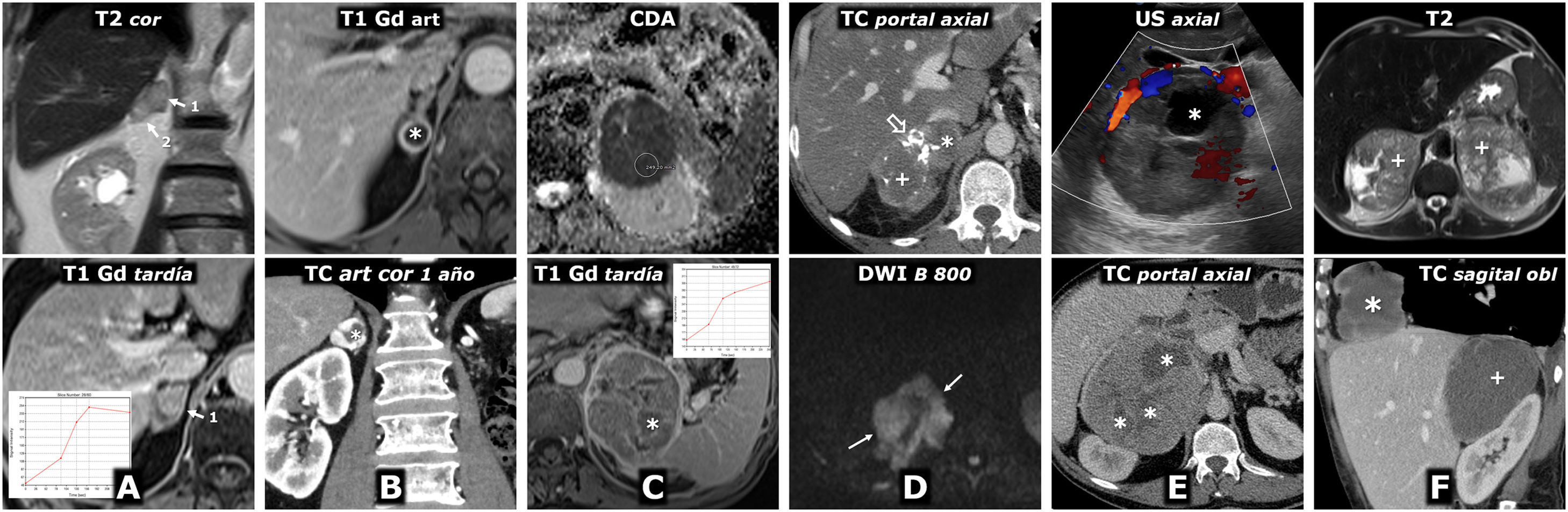
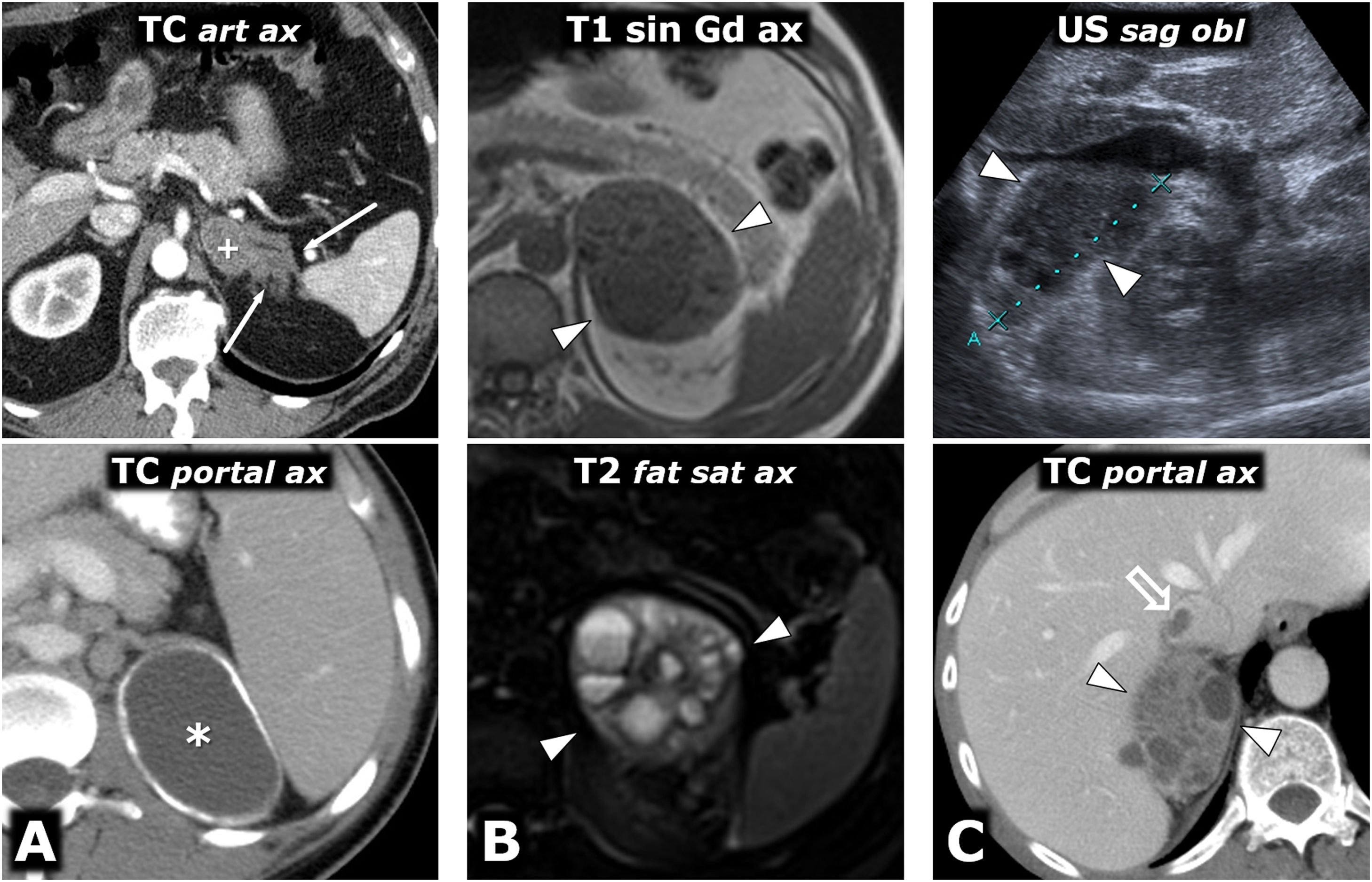
 Metástasis de carcinoma epidermoide de pulmón. Varón de 72 años. RM: hay 2 nódulos adrenales derechos sólidos (1 y 2), con moderadamente alta señal en T2, marcada restricción de la difusión del agua (no mostrado), sin lípidos microscópicos (no mostrado), con realce relativamente intenso, aunque algo más tardío de lo que es habitual en el feocromocitoma. B) Metástasis de carcinoma renal de células claras. Varón de 68 años intervenido 6 años antes de un carcinoma de células claras renal izquierdo. Nódulo adrenal derecho casi completamente quístico (*) con halo fino sólido con intenso realce en fase arterial y difusión libre del agua en su contenido (no mostrado). Un año después la lesión ha crecido y muestra en TC mayor componente sólido, con realce muy intenso en fase arterial. Nótese la ausencia de riñón izquierdo por nefrectomía. El aspecto radiológico aislado de la lesión en cualquiera de ambos estudios es indistinguible del de un feocromocitoma. C) Carcinoma adrenal primario. Varón de 67 años. RM: masa adrenal izquierda grande, bien delimitada, discretamente lobulada y con una estructura interna heterogénea. Las regiones sólidas no presentan caída significativa de su señal en fase opuesta, muestran hiperintensidad en T2 (no mostrado), muy marcada restricción de la difusión del agua (CDA de 0,61 x 10–3 mm2/s) y realce discreto (relativo menor del 100%) y progresivo. Hay focos necróticos y hemorrágicos (*) constatados histológicamente. D) Linfoma adrenal primario. Varón de 51 años VIH positivo. Masa adrenal derecha lobulada y muy heterogénea. Contiene calcificaciones (flecha hueca), áreas quísticas (*) y hemorrágicas (+), estas últimas con alta señal en T1 y halo hipointenso de hemosiderina en T2 (no mostrado). Las áreas sólidas presentan realce discreto en TC y RM (no mostrado) e intensa restricción de la difusión del agua, con alta señal con B alto (flechas) y valores muy bajos de CDA (no mostrado), en torno a 0,68 x 10–3 mm2/s. El diagnóstico histológico fue de linfoma B difuso de célula grande de inmunofenotipo activado (no GCB) asociado al virus de Epstein-Barr. E) Leiomiosarcoma pleomórfico adrenal. Varón de 40 años. Gran masa predominantemente sólida, aunque con áreas quísticas (*), lobulada y ejerciendo importante compresión en la vena cava inferior, que en la cirugía aparecía infiltrada. F) Angiosarcoma con afectación adrenal bilateral (+) y una tercera masa en el ángulo cardiofrénico derecho (*). Varón de 53 años. Hay 2 masas adrenales y una supradiafragmática, bien delimitadas, con baja densidad en TC, escaso realce casi solo periférico y en algunos septos. En RM presentan focos quísticos de diversos tamaños, algún pequeño foco hemorrágico y una importante restricción de la difusión del agua, con valores muy bajos de CDA (no mostrado). Se planteó la posibilidad de feocromocitoma en el contexto de un síndrome genético, con una tercera lesión que podría ser metastásica o corresponder a otro tumor. El estudio de catecolaminas fue normal. Se realizó BAG de una de las masas adrenales y el diagnóstico histológico fue de angiosarcoma epitelioide. Es imposible determinar cuál es el tumor primario.")
Diagnóstico diferencial. Lesiones adrenales malignas. A) Metástasis de carcinoma epidermoide de pulmón. Varón de 72 años. RM: hay 2 nódulos adrenales derechos sólidos (1 y 2), con moderadamente alta señal en T2, marcada restricción de la difusión del agua (no mostrado), sin lípidos microscópicos (no mostrado), con realce relativamente intenso, aunque algo más tardío de lo que es habitual en el feocromocitoma. B) Metástasis de carcinoma renal de células claras. Varón de 68 años intervenido 6 años antes de un carcinoma de células claras renal izquierdo. Nódulo adrenal derecho casi completamente quístico (*) con halo fino sólido con intenso realce en fase arterial y difusión libre del agua en su contenido (no mostrado). Un año después la lesión ha crecido y muestra en TC mayor componente sólido, con realce muy intenso en fase arterial. Nótese la ausencia de riñón izquierdo por nefrectomía. El aspecto radiológico aislado de la lesión en cualquiera de ambos estudios es indistinguible del de un feocromocitoma. C) Carcinoma adrenal primario. Varón de 67 años. RM: masa adrenal izquierda grande, bien delimitada, discretamente lobulada y con una estructura interna heterogénea. Las regiones sólidas no presentan caída significativa de su señal en fase opuesta, muestran hiperintensidad en T2 (no mostrado), muy marcada restricción de la difusión del agua (CDA de 0,61 x 10–3 mm2/s) y realce discreto (relativo menor del 100%) y progresivo. Hay focos necróticos y hemorrágicos (*) constatados histológicamente. D) Linfoma adrenal primario. Varón de 51 años VIH positivo. Masa adrenal derecha lobulada y muy heterogénea. Contiene calcificaciones (flecha hueca), áreas quísticas (*) y hemorrágicas (+), estas últimas con alta señal en T1 y halo hipointenso de hemosiderina en T2 (no mostrado). Las áreas sólidas presentan realce discreto en TC y RM (no mostrado) e intensa restricción de la difusión del agua, con alta señal con B alto (flechas) y valores muy bajos de CDA (no mostrado), en torno a 0,68 x 10–3 mm2/s. El diagnóstico histológico fue de linfoma B difuso de célula grande de inmunofenotipo activado (no GCB) asociado al virus de Epstein-Barr. E) Leiomiosarcoma pleomórfico adrenal. Varón de 40 años. Gran masa predominantemente sólida, aunque con áreas quísticas (*), lobulada y ejerciendo importante compresión en la vena cava inferior, que en la cirugía aparecía infiltrada. F) Angiosarcoma con afectación adrenal bilateral (+) y una tercera masa en el ángulo cardiofrénico derecho (*). Varón de 53 años. Hay 2 masas adrenales y una supradiafragmática, bien delimitadas, con baja densidad en TC, escaso realce casi solo periférico y en algunos septos. En RM presentan focos quísticos de diversos tamaños, algún pequeño foco hemorrágico y una importante restricción de la difusión del agua, con valores muy bajos de CDA (no mostrado). Se planteó la posibilidad de feocromocitoma en el contexto de un síndrome genético, con una tercera lesión que podría ser metastásica o corresponder a otro tumor. El estudio de catecolaminas fue normal. Se realizó BAG de una de las masas adrenales y el diagnóstico histológico fue de angiosarcoma epitelioide. Es imposible determinar cuál es el tumor primario.
Solo un 2-3% de las lesiones nodulares adrenales halladas incidentalmente son malignas.
- –
Metástasis. Diversos factores pueden dificultar el diagnóstico diferencial entre metástasis y feocromocitoma en un contexto neoplásico: valor limitado de la secuencia de difusión, posibles fenómenos quístico-necróticos en metástasis, solapamientos en patrón de realce en casos como tumor neuroendocrino o carcinoma renal de células claras y notable actividad metabólica de feocromocitomas no agresivos (fig. 4). La multiplicidad o la existencia de enfermedad diseminada pueden ser de utilidad en el diagnóstico diferencial.
- –
Carcinoma. Es infrecuente, con una prevalencia de 1-2 por millón. En general se comporta como una masa grande, lobulada y heterogénea, con áreas necróticas, quísticas y hemorrágicas, comportamiento infiltrante, extensión metastásica y realce más tardío y menos intenso que el del feocromocitoma. Las áreas sólidas presentan gran restricción de la difusión del agua.
- –
Linfoma. Casi siempre es secundario (en autopsias hasta en el 25% de los linfomas) y de diagnóstico sencillo en un contexto de amplia afectación retroperitoneal característica. El linfoma adrenal primario es excepcional y no es posible su diagnóstico radiológico con seguridad. Suele ser heterogéneo, con cambios quístico-hemorrágicos que pueden simular feocromocitoma, aunque con marcada restricción de la difusión y realce discreto en áreas sólidas.
- –
Sarcoma. Son excepcionales. El origen de los leiomiosarcomas adrenales primarios no se encuentra claramente determinado. Se considera que, como el resto de los leiomiosarcomas retroperitoneales, podrían originarse en la capa muscular de estructuras venosas (la vena adrenal o sus afluentes). Todos se comportan en general como masas adrenales grandes, lobuladas y heterogéneas, con áreas quísticas y necróticas, algunos con comportamiento más infiltrante.
- –
Adenoma. Es característica la caída de señal en fase opuesta hasta cierto rango, poco frecuente en feocromocitomas. Sin embargo, un 35% de estos presentan un lavado tardío similar en la TC (fig. 6) y un pequeño porcentaje de adenomas pueden tener cambios quísticos o hemorrágicos que planteen la posibilidad de feocromocitoma.
- –
Mielolipoma. El diagnóstico suele ser sencillo por la demostración de grasa macroscópica en TC o RM. Sin embargo, existe un pequeño porcentaje de mielolipomas con muy escaso componente de grasa, en los que puede ser más dificultoso.
- –
Hemangioma. Son raros (1/10.000 autopsias), predominando los cavernosos. Son lesiones bien delimitadas de pequeño tamaño con comportamiento similar al de los hemangiomas hepáticos: alta señal en secuencia T2 que puede simular feocromocitoma, pero realce globular periférico con progresión centrípeta, ocasionalmente incompleta. Pueden presentar flebolitos.
- –
Otros. Oncocitoma, schwannoma, ganglioneuroma, tumor adenomatoide. Muy raros. Todos pueden contener áreas quísticas.
 Adenoma con áreas focales quísticas. Mujer de 79 años. TC ante la sospecha de síndrome constitucional: lesión nodular con áreas quísticas (*) en la adrenal izquierda. En la RM, que no podemos mostrar al perderse las imágenes en un lamentable incidente informático, se apreciaba un área sólida con pérdida significativa de señal en fase opuesta, lo que sugería adenoma con cambios quísticos. Siete años después la lesión no se ha modificado en 2 controles de TC. No hay datos clínicos ni analíticos de feocromocitoma y no se ha diagnosticado ninguna neoplasia. B) Mielolipoma con escasa cantidad de grasa macroscópica. Hallazgo incidental en varón de 42 años en estudio por dolor abdominal. El nódulo adrenal (*) presenta alta señal en T2 y realce intenso y rápido en la mayor parte de la lesión, con escasa restricción de la difusión del agua (no mostrado). La clave diagnóstica la da la existencia de un foco excéntrico en la parte superior (flechas) que pierde señal en secuencias con saturación espectral de la grasa y STIR, por lo tanto, en relación con grasa macroscópica. C) Hemangioma adrenal. Mujer de 43 años. Hallazgo incidental de masa adrenal derecha bien delimitada (flechas), con alguna calcificación redondeada en TC sin contraste (flecha hueca). Muestra escasa restricción de la difusión del agua, con intensidad de señal relativamente baja, aunque heterogénea, con B alto, partiendo de una señal muy alta en T2 (no mostrado). En el estudio dinámico con contraste extracelular presenta un relleno progresivo y centrípeto siguiendo el pool vascular. Se confirmó histológicamente la sospecha radiológica de hemangioma.")
Diagnóstico diferencial. Lesiones adrenales benignas sólidas. A) Adenoma con áreas focales quísticas. Mujer de 79 años. TC ante la sospecha de síndrome constitucional: lesión nodular con áreas quísticas (*) en la adrenal izquierda. En la RM, que no podemos mostrar al perderse las imágenes en un lamentable incidente informático, se apreciaba un área sólida con pérdida significativa de señal en fase opuesta, lo que sugería adenoma con cambios quísticos. Siete años después la lesión no se ha modificado en 2 controles de TC. No hay datos clínicos ni analíticos de feocromocitoma y no se ha diagnosticado ninguna neoplasia. B) Mielolipoma con escasa cantidad de grasa macroscópica. Hallazgo incidental en varón de 42 años en estudio por dolor abdominal. El nódulo adrenal (*) presenta alta señal en T2 y realce intenso y rápido en la mayor parte de la lesión, con escasa restricción de la difusión del agua (no mostrado). La clave diagnóstica la da la existencia de un foco excéntrico en la parte superior (flechas) que pierde señal en secuencias con saturación espectral de la grasa y STIR, por lo tanto, en relación con grasa macroscópica. C) Hemangioma adrenal. Mujer de 43 años. Hallazgo incidental de masa adrenal derecha bien delimitada (flechas), con alguna calcificación redondeada en TC sin contraste (flecha hueca). Muestra escasa restricción de la difusión del agua, con intensidad de señal relativamente baja, aunque heterogénea, con B alto, partiendo de una señal muy alta en T2 (no mostrado). En el estudio dinámico con contraste extracelular presenta un relleno progresivo y centrípeto siguiendo el pool vascular. Se confirmó histológicamente la sospecha radiológica de hemangioma.
)
 Hematoma y seudoquiste. Imagen superior: varón de 63 años. Accidente de tráfico. Hemorragia adrenal izquierda traumática. Lesión nodular densa (58 UH) en la adrenal izquierda (+), con tractos densos hemáticos hacia la grasa perirrenal (flechas). En un estudio posterior se había convertido en un pequeño seudoquiste (no mostrado). Imagen inferior: varón de 42 años. Seudoquiste adrenal. Hallazgo casual de una masa quística adrenal izquierda con calcificación periférica (*) en un paciente en estudio por un síndrome linfoproliferativo (véanse la esplenomegalia y una pequeña adenopatía paraórtica izquierda) con antecedente traumático remoto. B) Quiste hidatídico. Varón de 56 años con HTA refractaria e insuficiencia renal (estudio sin contraste). Masa multiquística dependiente de la adrenal izquierda. No existía enfermedad hidatídica hepática ni en otras localizaciones. Pese a que en nuestra área de salud son aún frecuentes los casos de hidatidosis, se sospechó inicialmente feocromocitoma. Los estudios de catecolaminas fueron normales. El paciente fue sometido a intervención quirúrgica sin incidencias, con el resultado histológico de quiste hidatídico. C) Tuberculosis. Varón de 57 años, bebedor, con diagnóstico reciente de tuberculosis pulmonar (no mostrado). Se identifica una masa adrenal derecha multiquística (cabezas de flecha), con una pequeña extensión a la vena cava inferior (flecha hueca). Se realizó PAAF de esta lesión, con confirmación microbiológica de tuberculosis.")
Diagnóstico diferencial. Lesiones adrenales benignas quísticas. A) Hematoma y seudoquiste. Imagen superior: varón de 63 años. Accidente de tráfico. Hemorragia adrenal izquierda traumática. Lesión nodular densa (58 UH) en la adrenal izquierda (+), con tractos densos hemáticos hacia la grasa perirrenal (flechas). En un estudio posterior se había convertido en un pequeño seudoquiste (no mostrado). Imagen inferior: varón de 42 años. Seudoquiste adrenal. Hallazgo casual de una masa quística adrenal izquierda con calcificación periférica (*) en un paciente en estudio por un síndrome linfoproliferativo (véanse la esplenomegalia y una pequeña adenopatía paraórtica izquierda) con antecedente traumático remoto. B) Quiste hidatídico. Varón de 56 años con HTA refractaria e insuficiencia renal (estudio sin contraste). Masa multiquística dependiente de la adrenal izquierda. No existía enfermedad hidatídica hepática ni en otras localizaciones. Pese a que en nuestra área de salud son aún frecuentes los casos de hidatidosis, se sospechó inicialmente feocromocitoma. Los estudios de catecolaminas fueron normales. El paciente fue sometido a intervención quirúrgica sin incidencias, con el resultado histológico de quiste hidatídico. C) Tuberculosis. Varón de 57 años, bebedor, con diagnóstico reciente de tuberculosis pulmonar (no mostrado). Se identifica una masa adrenal derecha multiquística (cabezas de flecha), con una pequeña extensión a la vena cava inferior (flecha hueca). Se realizó PAAF de esta lesión, con confirmación microbiológica de tuberculosis.
- –
Quiste adrenal. Endoteliales (algunos autores incluyen los linfangiomas) o epiteliales. Se presentan como masas quísticas bien delimitadas de pared fina, ocasionalmente multiseptadas.
- –
Hematoma-seudoquiste. Ocasionados por traumatismo cerrado (más frecuentemente derecho), estrés o situaciones de gravedad (a menudo bilateral). En fase aguda se presenta como una lesión nodular de alta atenuación basal con áreas periféricas mal delimitadas. Al evolucionar suele mostrar una reducción progresiva de la atenuación y mejor delimitación hasta conformar un seudoquiste, habitualmente con calcificación periférica.
- –
Quiste hidatídico. Es excepcional, aunque debe tenerse en cuenta en áreas endémicas. Puede coexistir o no con enfermedad hepática o en otras localizaciones. Se presenta como lesión multiquística. Cuando se reconocen claras vesículas hijas o membranas el diagnóstico es más sencillo.
- –
Tuberculosis. La afectación adrenal es infrecuente. Suele ser bilateral, en forma de engrosamiento glandular con áreas quísticas o necróticas centrales y realce periférico. Puede ocasionar insuficiencia adrenal y devenir en calcificaciones residuales.
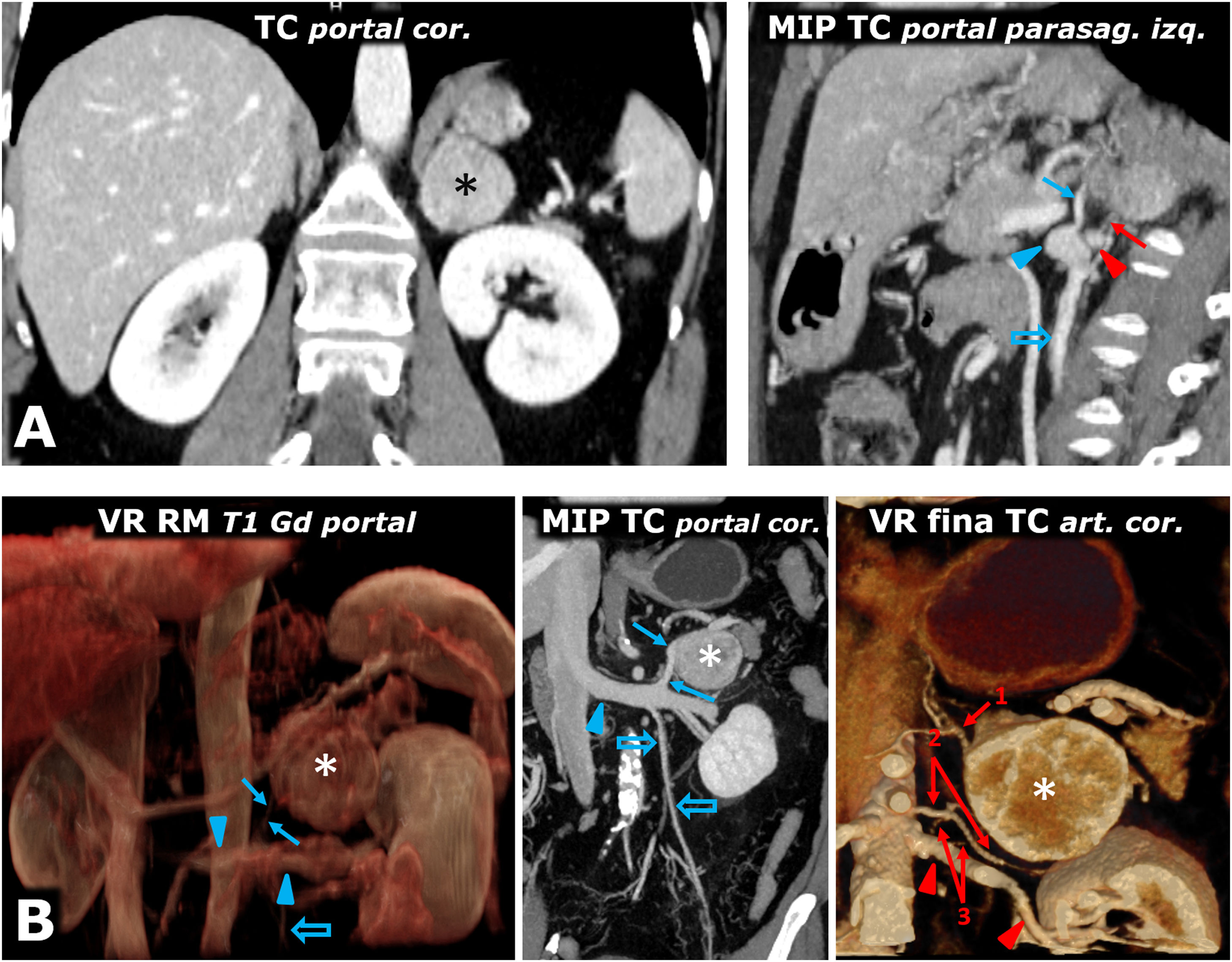
El tratamiento indicado es la resección quirúrgica completa, idealmente laparoscópica, que debe realizarse con un bloqueo adrenérgico previo. El radiólogo debe participar en la planificación3,5–8,12,14,24,30, revisando conjuntamente la anatomía vascular (especialmente venosa) y aportando reconstrucciones útiles (fig. 15). En casos de síndrome hereditario debe intentarse la resección conservadora de glándula. Conforme avanza la caracterización genética y molecular se van desarrollando terapias radiometabólicas, locales o sistémicas a aplicar individualizadamente7,8,12,14.
 Mujer de 57 años. Hallazgo incidental en ecografía (no mostrada) por hematuria de masa adrenal izquierda sólida (*) bien delimitada de 38mm en la adrenal izquierda, con realce intenso y lavado tardío similar al del adenoma (no mostrado). El estudio de catecolaminas fue patológico, decidiéndose intervención. Poco antes de la cirugía laparoscópica se revisa con los cirujanos la anatomía vascular, localizando las arterias adrenales superior (casi siempre rama de la frénica inferior), media (normalmente rama directa de la aorta) e inferior (flecha roja; en general rama de arteria renal ipsolateral), así como la vena adrenal (flecha azul; normalmente única y afluente de la vena renal en el lado izquierdo y de la cava inferior en el derecho) y todos los detalles anatómicos pertinentes. Cabeza de flecha roja: arteria renal izquierda. Cabeza de flecha azul: vena renal izquierda. Flecha hueca azul: vena ovárica izquierda. Se realizó resección laparoscópica sin incidencias. El tumor tenía criterios histológicos de agresividad (puntuación de 6 en la escala PASS). B) Varón de 73 años con HTA grado II. La reconstrucción volumétrica de la RM muestra las relaciones entre el feocromocitoma adrenal izquierdo (*), la vena adrenal (flecha azul) y la vena renal izquierda en la que desemboca (cabezas de flecha azul). Flecha hueca azul: vena espermática. Las reconstrucciones con TC aportan detalles anatómicos más precisos para la planificación quirúrgica. Flechas rojas: arterias adrenales izquierdas: 1: superior; 2: media, y 3: inferior (2 arterias como variante anatómica). Cabezas de flecha rojas: arteria renal izquierda.")
Ayuda en la planificación quirúrgica. A) Mujer de 57 años. Hallazgo incidental en ecografía (no mostrada) por hematuria de masa adrenal izquierda sólida (*) bien delimitada de 38mm en la adrenal izquierda, con realce intenso y lavado tardío similar al del adenoma (no mostrado). El estudio de catecolaminas fue patológico, decidiéndose intervención. Poco antes de la cirugía laparoscópica se revisa con los cirujanos la anatomía vascular, localizando las arterias adrenales superior (casi siempre rama de la frénica inferior), media (normalmente rama directa de la aorta) e inferior (flecha roja; en general rama de arteria renal ipsolateral), así como la vena adrenal (flecha azul; normalmente única y afluente de la vena renal en el lado izquierdo y de la cava inferior en el derecho) y todos los detalles anatómicos pertinentes. Cabeza de flecha roja: arteria renal izquierda. Cabeza de flecha azul: vena renal izquierda. Flecha hueca azul: vena ovárica izquierda. Se realizó resección laparoscópica sin incidencias. El tumor tenía criterios histológicos de agresividad (puntuación de 6 en la escala PASS). B) Varón de 73 años con HTA grado II. La reconstrucción volumétrica de la RM muestra las relaciones entre el feocromocitoma adrenal izquierdo (*), la vena adrenal (flecha azul) y la vena renal izquierda en la que desemboca (cabezas de flecha azul). Flecha hueca azul: vena espermática. Las reconstrucciones con TC aportan detalles anatómicos más precisos para la planificación quirúrgica. Flechas rojas: arterias adrenales izquierdas: 1: superior; 2: media, y 3: inferior (2 arterias como variante anatómica). Cabezas de flecha rojas: arteria renal izquierda.
Es posible la recidiva local tras la resección quirúrgica, incluso muchos años después. Se recomienda el seguimiento a largo plazo, durante al menos 10 años tras la operación y en ciertas situaciones de por vida3,6–8.
ConclusionesAunque la presentación clínica y las alteraciones analíticas pueden orientar el diagnóstico de feocromocitoma, no es infrecuente que sea el radiólogo el primero en sugerirlo por los hallazgos en una prueba de imagen.
Debe recordarse que la presentación característica es una lesión nodular adrenal bien delimitada que presenta alta señal en T2, realce rápido e intenso y con frecuencia fenómenos quísticos y hemorrágicos.
En casos dudosos, puede recomendarse la determinación de catecolaminas o exploraciones complementarias de Medicina Nuclear.
Debe evitarse la punción percutánea por las posibles complicaciones secundarias, potencialmente graves.
Pueden verse feocromocitomas en varios síndromes hereditarios, sobre todo von Hippel-Lindau, MEN 2 y paraganglioma hereditario. En casos esporádicos, subyacen también muy frecuentemente alteraciones genéticas que están cambiando el paradigma de la enfermedad.
Han de conocerse las numerosas lesiones con las que se puede plantear el diagnóstico diferencial.
Autoría- 1.
Responsable de la integridad del estudio: MACC.
- 2.
Concepción del estudio: MACC.
- 3.
Diseño del estudio: MACC.
- 4.
Obtención de los datos: MACC, JEI, GCFP, MRC y AF.
- 5.
Análisis e interpretación de los datos: MACC, JEI, GCFP, MRC y AF.
- 6.
Tratamiento estadístico: no procede.
- 7.
Búsqueda bibliográfica: MACC y JEI.
- 8.
Redacción del trabajo: MACC, JEI y GCFP.
- 9.
Revisión crítica del manuscrito con aportaciones intelectualmente relevantes: MACC, JEI, GCFP, MRC y AF.
- 10.
Aprobación de la versión final: MACC, JEI, GCFP, MRC y AF.
Los autores declaran no tener ningún conflicto de intereses.

