



En esta parte de la revisión se describe la relación funcional entre el metabolismo de los lípidos y los hidratos de carbono y su interdependencia, desde el ciclo glucosa-ácido grasos y la hipótesis portal de la insulinorresistencia a los nuevos conocimientos sobre los adipocitos marrones y beiges, con énfasis en el normal funcionamiento de un patrón endocrino cuya disfunción es clave en la fisiopatología de la DMT2 y la obesidad. Se discute la ectopia o el asiento de grasa en el tejido magro por incapacidad del tejido adiposo para seguir acopiando lípidos y la actividad endocrina del adipocito, con la producción de moléculas (adipoquinas) que influyen sobre los mecanismos inductores de insulinorresistencia (leptina, adiponectina, TNF-α, resistina, etc.) y disfunción de la célula beta. Se describen la disminución de la capacidad oxidativa en la cadena respiratoria mitocondrial y el renacer del concepto de lipogénesis de novo, ambas favoreciendo el acopie de lípido intracelular. En tejidos magros existen pequeñas reservas intracelulares de lípidos que mantienen la regulación de funciones esenciales, aunque si aparece una sobrecarga lipídica el fenómeno conduciría a una disfunción (lipotoxicidad) y a la muerte celular (lipoapoptosis). La tormentosa relación entre los lípidos y el islote de Langerhans va más allá del esfuerzo funcional que impone la insulinorresistencia periférica sobre la célula β, por efectos directos de los lípidos o de sus derivados sobre la función del islote pancreático. Sin déficit de insulina no se desarrolla diabetes.
In this part of the review, the functional relationship between lipid and carbohydrate metabolisms and their interdependence is described, from the glucose-fatty acid cycle and the portal hypothesis of insulin resistance to the new knowledge on brown and beige adipocytes, with emphasis on the normal functioning of an endocrine pattern in which its dysfunction is a key factor in the pathophysiology of T2DM and obesity. Ectopic fat deposition in lean tissues due to the inability of the adipose tissue to continuously collect lipids and the endocrine activity of adipocytes is discussed. The production of molecules (adipokines) influencing some of the mechanisms involved in the development of insulin resistance (leptin, adiponectin, TNF-α, resistin, etc.) and beta cell dysfunction is also revisited. The decrease in the oxidative capacity in the mitochondrial respiratory chain and the rebirth of the concept of de novo lipogenesis are described, both effects favouring intracellular lipid accumulation. In lean tissues there are small intracellular lipid reserves that help to maintain the regulation of essential functions; however, when a lipid overload occurs the phenomenon could lead to severe cell dysfunction (lipotoxicity), and death (lipo-apoptosis). The stormy relationship between lipids and the Langerhans’ islets goes beyond the functional effort imposed by peripheral insulin-resistance on the β cells, either by the direct effect of lipids or by their derivatives on overall pancreatic islet function. Within a scenario of no insulin deficit, diabetes does not develop.
La epidemia de obesidad y diabetes mellitus tipo 2 (DMT2) tiene en realidad un sutil integrante: la insulinorresistencia. Dado que el crecimiento asombroso de estos problemas condiciona la salud pública mundial, son las afecciones metabólicas las que matan a través de la enfermedad cardiovascular. Las investigaciones han sido esenciales para desarrollar la idea de asociación entre la obesidad y la DMT2. Así, progresivamente no solo se vinculó a la severidad de la obesidad, sino a la ganancia de peso corporal, la duración de tal ganancia, el tipo clínico y también la evidencia de la prevención o mejora del trastorno metabólico cuando la persona obesa adelgaza.
La insulinorresistencia como componente esencial en la etiopatogenia de la DMT2 se ha definido tradicionalmente desde el punto de vista glucotóxico. Con el pasar de los años se gestó y se fortaleció la idea de que las grasas influyen de manera decisiva sobre el metabolismo de los hidratos de carbono, la actividad de la insulina y, a través de ellos, en el desarrollo de la DMT2. Existen numerosos trabajos que cimentaron una interesante historia que, en parte, muestra la tormentosa relación entre la obesidad y la diabetes; los ácidos grasos (AG) libres (AGL) y la glucemia, y el tejido adiposo y los islotes de Langerhans.
En los últimos años ha cambiado dramáticamente la opinión que atribuía a los adipocitos la característica de conformar un tejido para asegurar la reserva energética. Se han hallado y se les atribuye tantas actividades que es difícil reunir todas sus funciones, que los muestra además como una verdadera glándula endocrina1 con un desempeño vital en el metabolismo. El adipocito como órgano dinámico regula finamente el balance nutricional a través y por medio de un complejo intercambio con los órganos y el medio que lo rodea, desarrollando entonces una actividad endocrina-paracrina-autocrina.
Existen defectos génicos y efectos ambientales capaces de inducir desde acumulación de grasa central abdominal y en tejidos no adiposo (depósito ectópico de grasa), hasta la movilización de lípidos y la producción de diversas sustancias que se originan en el tejido adiposo pero que influyen en la función de otros órganos, todo como reflejo de un fenómeno de mal adaptación del metabolismo energético. Tal vez, los cambios en los lípidos séricos y tisulares se constituyen en los mayores perpetuadores de la insulinorresistencia. No se conoce aún cuál es el fenómeno original, pero tradicionalmente se considera que las alteraciones en el intercambio de glucosa y de AGL en el músculo y la pérdida de la capacidad para suprimir la liberación de AGL y glicerol desde los adipocitos son fenómenos tempranos en la disfunción metabólica2-4.
Este conjunto de anomalías nos ha llevado a la revisión de aspectos esenciales del vínculo entre los lípidos y la DMT2: la influencia de los AGL, los conceptos sobre grasa central y ectópica, el paradigma del adipocito secretor y los hallazgos de novedosas evidencias sobre mecanismos que intervienen en la patogenia de la DMT2. Asimismo, es imperioso reconocer que el tejido adiposo es esencial en la regulación del gasto energético, por tanto, para la vida, con la propuesta ya analizada que dentro de la homeostasis existiría un eje adipo-insular. El problema es cuando se rompe el equilibrio y el adipocito normo-funcional pasa a ser disfuncional.
No solo el tejido adiposo blanco interesa: los adipocitos marrones y mioquinas tienen relevancia en la obesidad y la diabetesLas células adiposas se especializaron para preservar energía y pueden actuar de sensores que generan diferentes respuestas a los cambios en el metabolismo energético. Entre las investigaciones de los últimos años se destacaron 2 temas: 1) la biología de adipocitos diferentes, el blanco y el marrón, y 2) las moléculas reguladoras con influencia en la fisiología y la homeostasis energética de los animales (fig. 1).
. Adaptado de Spiegelman17.")
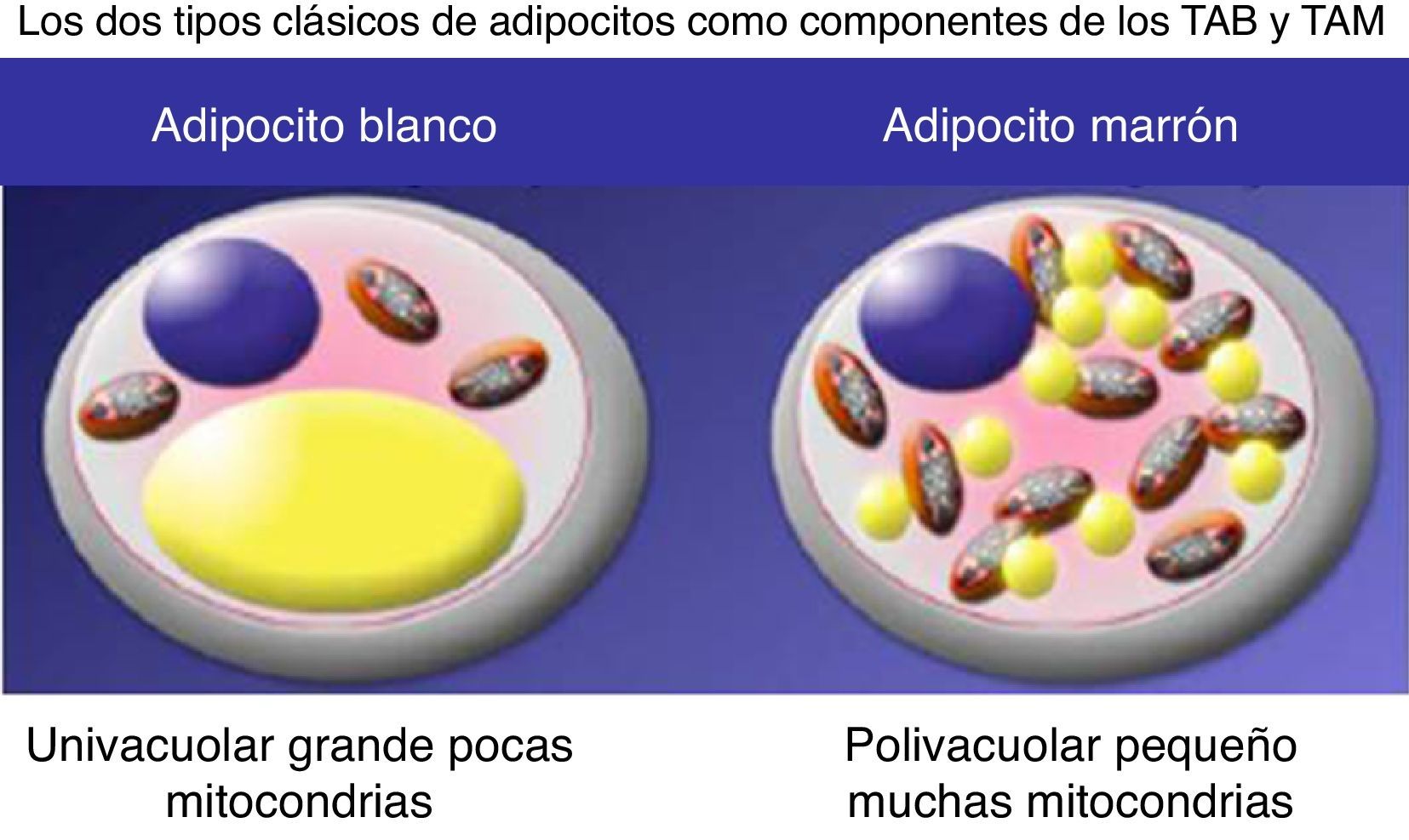
Características diferenciales de los adipocitos que conforman el tejido adiposo blanco y el marrón (o pardo).
Adaptado de Spiegelman17.
El adipocito blanco maduro almacena energía en una sola gota lipídica (univacuolar), tiene un contenido mitocondrial relativamente bajo y escasa proteína desacoplante-1 (UCP-1), produce moléculas de señal que influyen en la ingesta y la sensibilidad, y secreción de insulina. En cambio, el adipocito marrón maduro es polivacuolar (múltiples y pequeñas gotas de lípidos) y con un alto contenido de mitocondrias y UCP-1, disipando así la energía química en forma de calor (fig. 2).
. Este proceso disipador de energía ocurre a expensas del aumento en el número de mitocondrias intraadipocitarias. Adaptado de Gura T. Uncoupling proteins provide new clue to obesity")
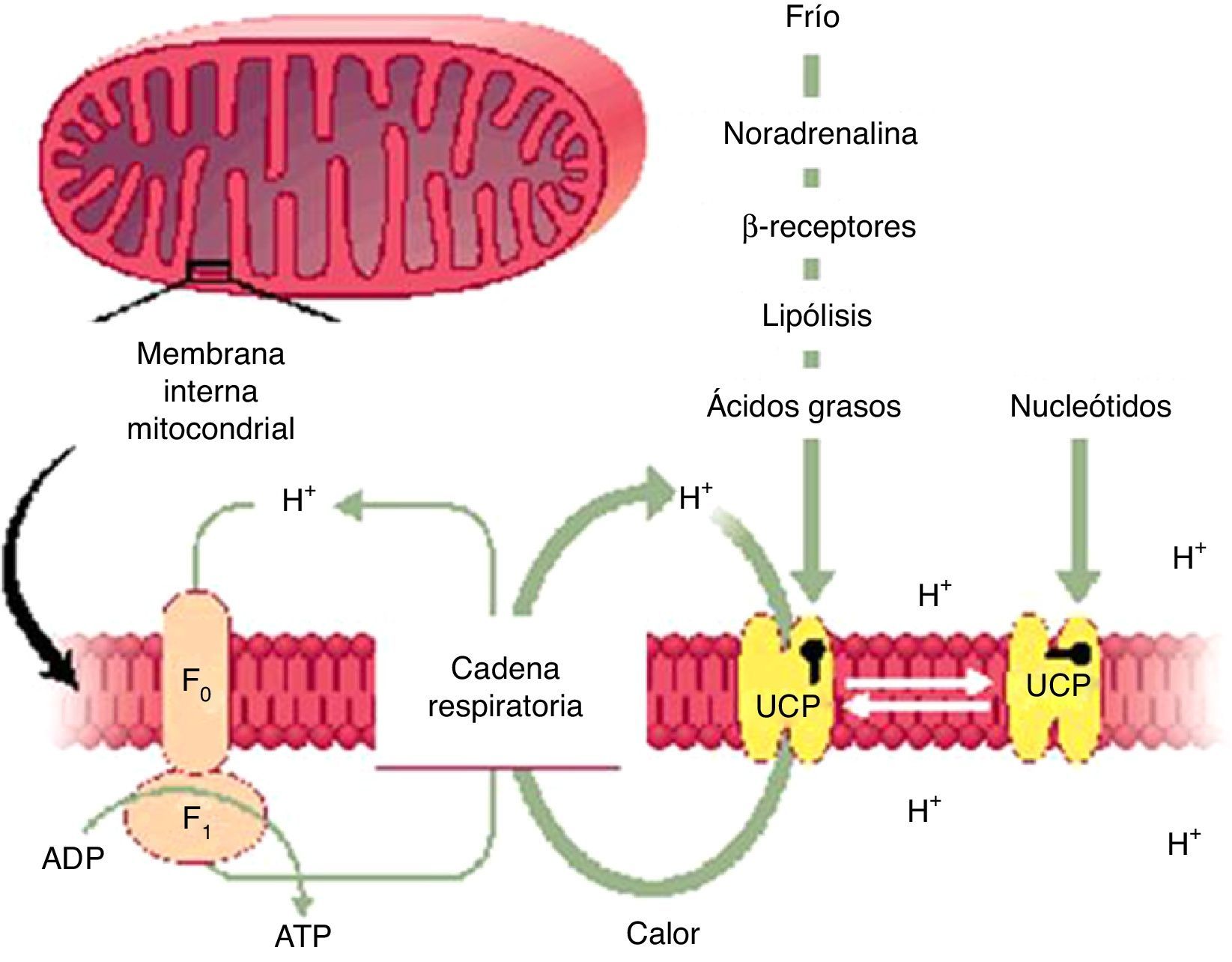
Esquema representativo del proceso mitocondrial termogénico que ocurre, fundamentalmente, en adipocitos marrones (y «amarronados»). Este proceso disipador de energía ocurre a expensas del aumento en el número de mitocondrias intraadipocitarias.
Adaptado de Gura T. Uncoupling proteins provide new clue to obesity's causes. Science. 1998 29;280:1369-70.
Así como se identificó que las células del tejido adiposo blanco (TAB) al hipertrofiarse en la obesidad, entre otros cambios, producen una moderada cantidad de sustancias proinflamatorias5, también se determinó que el PPAR-γ es uno de los factores reguladores dominantes en el desarrollo de células del tejido adiposo marrón (TAM)6. A partir de estos conocimientos, se desarrolló el concepto de que estas células, además de proteger de la hipotermia a la mayoría de los mamíferos, tienen una natural influencia antiobesidad (endógena) y también de resguardo contra la DMT2, por su alto contenido en mitocondrias y de UCP-1 (generan protones que filtran a través de la membrana interna de la mitocondria y disipan energía química en forma de calor). Los interrogantes claves sobre el TAM se dirigieron a conocer cuáles serían sus funciones dentro del balance energético global y además cómo son los mecanismos endógenos que regulan y modifican la cantidad y/o la función del TAM.
Si bien ambos tejidos adiposos requieren de la actividad PPAR-γ, el TAM necesita componentes moleculares adicionales para su desarrollo entre los que se incluyen las denominadas PGC17, PGC1-α y β-PGC1, y particularmente PRDM16, que pareció controlar el derrotero hacia el desarrollo a TAM8.
En el avance de conocimientos, el primer paso fue redescubrir la presencia del TAM en los seres humanos a través de la tomografía captadora de la emisión de positrones (se utilizaba en diagnósticos oncológicos).
Luego, se determinó que la actividad del PRDM16 es indispensable para el desarrollo de TAM. En cultivos de células precursoras de TAM (que finalizaban diferenciándose en adipocitos marrones), se suprimió el PRDM16 mediante un silenciador de la expresión de ARNm (shRNA), que anuló la función del gen específico y los precursores desarrollaron estructuras que contenían proteínas musculares. Así se demostró que la supresión de PRDM16 cambió la diferenciación de las células de grasa marrón a células del músculo esquelético. Los resultados se confirmaron por el camino inverso: la expresión de PRDM16 en mioblastos (por técnicas biotecnológicas) los diferenció a células del TAM9.
Los resultados indicaron que el TAM y el músculo esquelético derivan de un mismo linaje celular. En cambio, el TAB deriva de un linaje celular diferente9. Sin embargo, se evidenció algo sorprendente: en condiciones particulares (extremo frío o intensa actividad β-adrenérgica) en el TAB surgieron parches con adipocitos marrones, lo cual proporcionó la primera evidencia que habría 2 tipos de células diferentes a las que llamaba células del TAM:
- 1.
el clásico o típico, que se encuentra en los depósitos interescapular y perirrenal, y
- 2.
«beige o amarronadas», que emergen en la masa del TAB y contienen UCP-110.
Resta determinar aún, si hay subtipos adicionales de adipocitos que disipen la energía química.
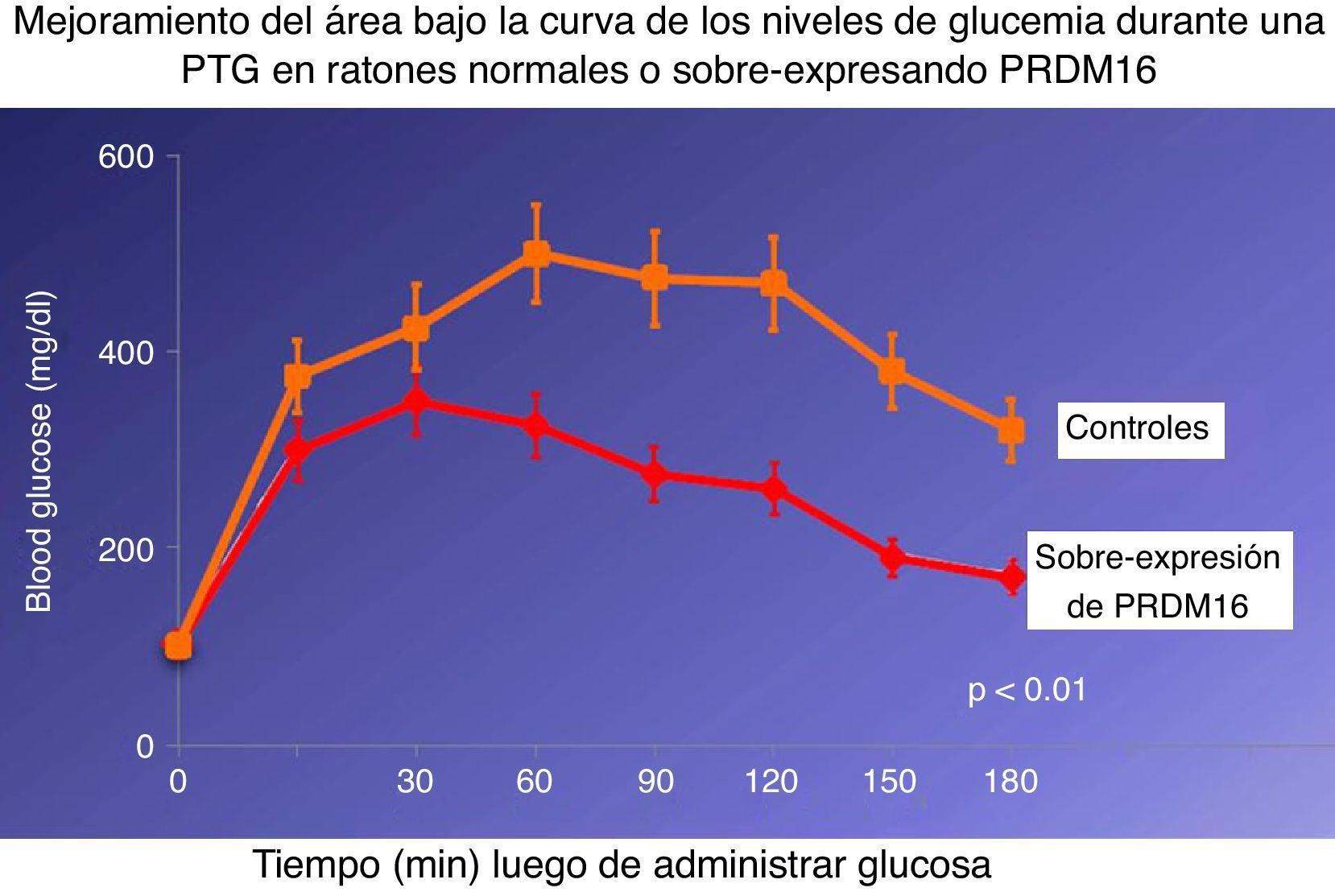
Los efectos metabólicos se investigaron en ratones control normales y en transgénicos con sobreexpresión (manipulación genética) de PRDM16 sin alteraciones en el tejido adiposo visceral ni en el TAM clásico, pero con una pardización en el TAB subcutáneo, a los que sometió a dieta hipergrasa11. Los ratones transgénicos tuvieron un área bajo de la curva de glucosa significativamente más pequeña que la de controles, que indicó una mejor tolerancia a la carga de glucosa (fig. 3). Se concluyó que la presencia de más grasa beige puede mejorar el metabolismo y la homeostasis de los hidratos de carbono.

Una sobreexpresión de la proteína PRDM16 en células precursoras del tejido adiposo marrón resulta en una mejor tolerancia a la sobrecarga con glucosa en un individuo adulto.
Adaptado de Spiegelman17.
Una respuesta sorprendente fue que el ejercicio (actividad física) indujo la pardización («browning») del TAB.
La molécula de PGC1-α, es un potente coactivador de transcripción y regulador clave de la biogénesis mitocondrial en mamíferos. Su presencia es mayor en TAM que en TAB, y cuando se incorpora en este se expresan más mitocondrias y algunos elementos (no todos) del proceso de «pardización» (amarronamiento).
La expresión celular de PGC1 se incrementa con el ejercicio en animales experimentales y en seres humanos (correr y nadar)12,13 y se produce biogénesis mitocondrial. Pero también estimula la captación de glucosa, la formación de la unión neuromuscular, la angiogénesis y la oxidación de AG14. Se accedió, en parte, a la compleja fisiología del ejercicio (múltiples variables) por un modelo experimental de células musculares de un animal transgénico con solo un cambio (p. ej., elevación del PGC1-α). Así, se observó que los ratones con expresión transgénica de PGC1 en el músculo tenían mayor proceso de pardización del TAB subcutáneo y de mRNA de UCP-1, visible en células teñidas del TAB inguinal15.
La pregunta sobre la posibilidad de una molécula muscular bajo la regulación de PGC1-α, que vincule las funciones de los tejidos muscular y adiposo, tuvo su respuesta al determinar experimentalmente una proteína soluble de membrana tipo 1 con un dominio extracelular, que provenía del tejido muscular que indujo la pardización del TAB, sin modificar la función del TAM clásico, se identificó como FNDC515 y afianzó la idea que la activación de la grasa marrón típica no es lo mismo que la pardización del TAB (FNDC5 es activa en un tipo de grasa y no en el otro).
La molécula de FNDC5 se clonó y se estudió en profundidad. Se registró como una proteína intracelular, con función y organización de péptido de señal con dominio de fibronectina tipo 3 y un tramo corto hidrófobo que semeja un dominio de transmembrana16. Se identificó el fragmento secretado por la escisión de FNDC5 como un péptido que representó los aminoácidos 30-143 (112 amino ácidos) y se consideró una mioquina a la que se denominó «irisina» (de Iris: diosa griega que tomó los mensajes de los principales dioses del Olimpo y los bajó a los humanos de la tierra).
Un aspecto intrigante de irisina es que es 100% de su estructura es idéntica en la mayoría de las especies de mamíferos, que circula en sangre en el orden de 50nM y actúa sobre las células del TAB subcutáneo para inducir «pardización», cuya actividad protege al organismo de enfermedades metabólicas. Debería enfatizarse que el ejercicio proporciona grandes beneficios a otros tejidos, tales como el cerebro, el hígado, el corazón y al propio músculo esquelético. Asimismo, se reconoce como uno de los factores sobresalientes que causa neurogénesis en seres humanos adultos. También se ha demostrado que lleva a cabo una cierta mejora en pacientes con enfermedad de Alzheimer, Parkinson y otras enfermedades neurodegenerativas.
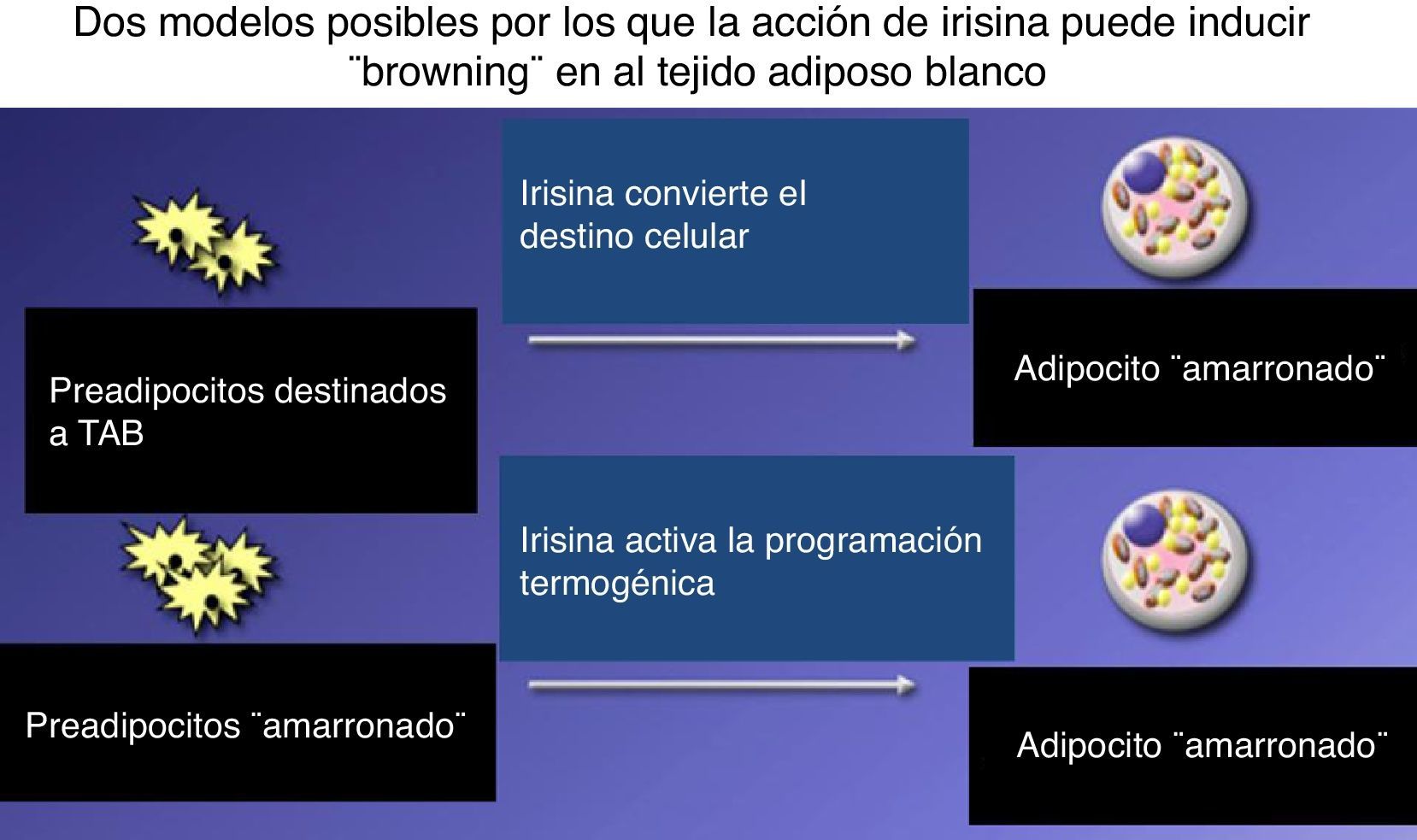
Se concluye que hay (por ahora) 2 tipos de células de TAM: las «clásicas» y las «beige o amarronadas», que aparecen dentro de áreas de TAB (fundamentalmente subcutáneo). El descubrimiento de la irisina implicó un mecanismo de señal: FNDC5 es una proteína de superficie que por proteólisis produce una mioquina de 112 aminoácidos que pasa a la circulación y actúa sobre preadipocitos específicos dentro del TAB (que responderían a estímulos para UCP1, a diferencia de los que quedan blancos, sin posibilidad para responder), modifica su desarrollo y pardiza las células al activar la programación celular para desarrollar su capacidad termogénica (fig. 4). La grasa termogénica del hombre semeja los adipocitos beiges de los roedores más que la clásica célula marrón15.
 para inducir el «amarronamiento» del tejido adiposo blanco, la irisina: podría convertir el destino de una célula progenitora de un adipocito blanco o (¿y?) podría activar la adipogénesis a partir de una célula progenitora de adipocito amarronado. Adaptado de Spiegelman17.")
Postulación alternativa del efecto de la mioquina (irisina) para inducir el «amarronamiento» del tejido adiposo blanco, la irisina: podría convertir el destino de una célula progenitora de un adipocito blanco o (¿y?) podría activar la adipogénesis a partir de una célula progenitora de adipocito amarronado.
Adaptado de Spiegelman17.
La irisina es un atractivo ejemplo del campo de la investigación traslacional, ya que la experimentación en biología molecular celular avizora su aplicación para la prevención de la obesidad y la DMT2 humana, y quizás también como potencial terapéutica para la obesidad17.
La grasa ectópica: ¿por qué y cómo los lípidos se guardan en lugares erróneos?Una gran parte de la preocupación del vínculo entre la disfunción adipocitaria y la DMT2 se refiere a que existen evidencias de que las grasas se sitúan en órganos y tejidos no preparados para su depósito, en vez de hacerlo en su lugar natural: el tejido adiposo. Esos depósitos se ubican dentro o alrededor de tejidos y órganos magros, como los músculos esqueléticos, el miocardio, los vasos, el hígado, el riñón y el páncreas. Por eso, el aumento del contenido de triglicéridos dentro del músculo esquelético entre los obesos y los diabéticos se considera un potente predictor de insulinorresistencia18.
La captación, la síntesis y la reserva de los triglicéridos de los adipocitos y la movilización de esta fuente de energía como AG se regulan por delicados mecanismos que integran factores genéticos, nutricionales y secreción endocrina y paracrina.
Evolutivamente, los adipocitos han servido al propósito de proteger la vida durante los ciclos de hambruna, ya que permiten reservar combustibles como triglicéridos en los momentos de disponibilidad, para utilizarlos en los periodos de necesidad de los tejidos y órganos magros (como de alguna manera lo expresó la hipótesis del «genotipo ahorrador de Neel»)19.
Las pequeñas reservas de grasas intracelulares de los órganos no adiposos se utilizan para esenciales funciones de las células, como la manutención de las membranas, la fluidez y las señales celulares. Pero el depósito de un modesto exceso de grasa dentro de un tejido magro provoca cambios clínicos, como resistencia a la insulina, hígado graso, cardiomiopatía y DMT2. Unger y McGarry, en 1994, acuñaron el término lipotoxicidad para denominar la disfunción que produce la presencia anormal de grasa en un órgano o tejido magro, y que incluso puede inducir a la muerte celular (lipoapoptosis)20.
Un balance graso positivo favorece el depósito adiposo. Esto puede provenir de la dieta, sea en forma directa como lípidos exógenos y/o como precursores no lipídicos que a través del proceso de “lipogénesis de novo” puden finalizar como AG. Para que esto último ocurra, se requieren sustratos (p. ej. hidratos de carbono) que cuando sean catabolizados generen AcetilCoA. Schutz admitió, que aún son incipientes los conocimientos sobre la lipogénesis de novo y la mayor parte de sus conceptos «se encuentran aún en la infancia»21.
Mientras el tejido adiposo conserve su facultad de atesoramiento de grasas, crecerá (en oportunidades en exceso: obesidad) y protegerá al resto de los órganos de la actividad lipotóxica («adipocito amigo»). Sin embargo, existe un cierto momento en el que se pierde la capacidad de reserva y los tejidos magros sufren la sobreoferta de grasas y sus consecuencias. Ese límite, que aún no se ha podido definir ni cuándo ni cómo se produce, se conoce como «umbral crítico» («adipocito enemigo»)22.
En la hipótesis del umbral crítico de adiposidad visceral, existe un rango individual para que se acumule una cantidad crítica de tejido adiposo visceral. La sensibilidad del adipocito a la insulina es importante para engordar y acumular masa grasa visceral23.
Una vez que se alcanza el umbral, disminuye la sensibilidad a la insulina y el adipocito en una actitud protectora, para que no se siga atesorando grasa, se hace insulinorresistente, con las consecuencias generales y las manifestaciones de síndrome metabólico y/o diabetes. Algo notable es que una modesta pérdida de peso (5-10%) se acompaña de una disminución del tejido adiposo visceral y un freno o reversión del proceso.
En la hipótesis se especula que el tejido adiposo es incapaz de mantener secuestradas las grasas para mantenerla fuera del hígado y el músculo. Cualquier perturbación que provoque la acumulación de AG-AcilCoA y otros metabolitos en estos tejidos provocaría resistencia a la insulina.
Una aparente paradoja la aportan los cuadros de lipodistrofia en humanos con disminución marcada del tejido adiposo, ya que se acompañan de diabetes y manifestaciones de insulinorresistencia. La falta de tejido adiposo en el que se puedan guardar conduce al exceso de depósito de grasas en tejidos magros y ello provoca la disminución de la sensibilidad a la insulina y la diabetes. Esta hipótesis encuentra apoyo a nivel experimental con ratones transgénicos que nacen sin tejido adiposo (lipoatrofia) y muestran el depósito de grasas en múltiples órganos e insulinorresistencia severa.
Moitra y su gran grupo de colaboradores estudiaron a un ratón cuyos adipocitos expresan la proteína AZIP/F-1, que bloquea la función de diversos factores de transcripción (interfiere la unión del DNA)24.
El ratón transgénico denominado A-ZIP/F-1 no tiene TAB y reduce marcadamente el TAM, el que, a su vez, es inactivo. Tienen un retardo del crecimiento pero en 12 semanas sobrepasan su peso habitual, comen, beben y orinan copiosamente, tienen una fecundidad disminuida y muerte prematura. Las consecuencias fisiológicas son profundas: muestra una severa insulinorresistencia, hígado graso y visceromegalia (triplican los triglicéridos hepáticos y musculares), es diabético (la glucemia se eleva por 3), presenta disminución de leptina (20 veces) y aumento de insulina (50 a 400 veces), AGL (por 2) y triglicéridos (por 3-5). El defecto en la acción hormonal es en IRS-1/IRS-2, que depende de la fosfoinositol 3 cinasa tanto en el músculo, como en el hígado.
Impresiona el hecho de que al trasplantar tejido adiposo normal al ratón A-ZIP/F1, por un lado, normaliza el contenido de lípidos en el hígado y el músculo, y la actividad de la insulina, aunque, por el otro, no se restauran por completo las alteraciones metabólicas. De alguna manera esto significa que la disminución de los triglicéridos secuestrados es insuficiente para que se logre la restauración total25.
Es notable como la obesidad con adipocitos «llenos» y disfuncionales tiene una repercusión metabólica similar a la falta de tejido graso, como sucede en la lipoatrofia (adipocito «ausente»).
El paradigma del adipocito «endocrino-paracrino-autocrino»Los adipocitos responden a diversas señales y estímulos endocrinos y neurales mediante la formación de proteínas (citoquinas, quimioquinas y hormonas) de las que algunas se vuelcan al torrente circulatorio como la interleucina-6 (IL-6), resistina, adiponectina y leptina. En cambio, otras actúan de manera paracrina o autocrina, como el factor de necrosis tumoral-α (TNF-α). Es probable, además, que el gran adipocito inmaduro tenga una producción y secreción de sustancias diferentes del adipocito pequeño y maduro.
La cualidad secretoria del adipocito lo vincula con diferentes funciones del organismo y con actividades críticas ocasionalmente duales o múltiples, como el apetito, el balance energético, el metabolismo y la sensibilidad a la insulina (AGL, adiponectina, resistina, agouti, ligandos de PPAR). Asimismo sobre la inmunidad (TNF-α, IL-6, complementos y proteína acetiladora), la angiogénesis, la presión arterial, la hemostasia (PAI-1, renina-angiotensina) y el sistema endocrino, e incluso también sobre la reproducción (leptina, corticoides, esteroides sexuales, visfatina)26.
Trayhurn consideró que estas proteínas no son estrictamente citoquinas y recomendó que se denomine adipoquinas a las sustancias que se sintetizan y secretan por el adipocito, y excluyó de esta calificación las proteínas que se producen en otros sitios del tejido adiposo, como por ejemplo en los macrófagos27.
Se han descubierto numerosas adipoquinas (tabla 1) pero esto constituye un tema «en progreso», ya que se continúan investigando y describiendo nuevos elementos y funciones28. Así, se ha demostrado la producción de diversas sustancias (no todas fabricadas por los adipocitos, sino incluso por otras estructuras que constituyen el tejido adiposo), entre las que se encuentran: leptina, adiponectina, resistina, omentina, PAI-1, quemerina, TNF-α, IL-6, visfatina y apelina, proteínas acetiladoras [ASP], etc.29.
Adipoquinas: sus funciones y relaciones recíprocas
| Adipoquina | Función metabólica | Efecto sobre otras adipoquinas |
|---|---|---|
| Adiponectina | Supresión de gluconeogénesis hepática | Supresión de la expresión de TNF-alfa y de IL-6 |
| Estímulo de oxidación de AG en el hígado y el músculo esquelético | ||
| Estímulo de la captación de glucosa por el músculo esquelético | ||
| Estímulo de la secreción de insulina | ||
| Modulación de la ingesta alimentaria y el gasto de energía | ||
| Leptina | Represión de la ingesta | Estimulación de la expresión de adiponectina |
| Promoción del gasto energético | Estimulación de la expresión de TNF-α e IL-6 | |
| Estimulación de oxidación de AG en hígado, páncreas y músculo | Supresión de la expresión de resistina y retinol | |
| Modulación de la gluconeogénesis hepática | ||
| Modulación de la célula beta del páncreas | ||
| Resistina | Inducción de insulinorresistencia en roedores | Estimulación expresión de TNF-α e IL-6 |
| Ausencia de evidencias de funciones en el metabolismo glúcido en humanos | ||
| Omentina | Mejora del transporte de glucosa estimulado por insulina | No se conoce |
| Fosforilación de Akt en adipocitos humanos | ||
| Quemerina | Mejora del transporte de glucosa estimulado por insulina | Supresión de la expresión de TNF-α y de IL-6 |
| Fosforilación de IRS-1 en adipocitos | ||
| PAI-1 | Inflamatoria y protrombótica | |
| TNF-α e IL-6 | Modulación de la señal de insulina en el hígado y el músculo esquelético | Estimulación de leptina, resistina, y visfatina |
La producción de citoadipoquinas antiinflamatorias y proinflamatorias, como IL-6 y el TNF-α, y de quimioquinas (una gran familia de pequeñas proteínas involucradas en la vigilancia inmunológica y en la activación y el reclutamiento de poblaciones celulares específicas durante la enfermedad) evidencia la participación del tejido adiposo en la respuesta inmunitaria innata y adquirida. Además, se considera que tanto la leptina como la adiponectina, la resistina y la visfatina participan en la regulación de la función de monocitos y macrófagos que secretan moléculas que se conectan con la respuesta inmunitaria innata. Asimismo, algunos autores consideran que los preadipocitos y adipocitos expresan un amplio espectro de receptores «toll-like», que pueden convertirlos en células símil-macrófagos30. No se descarta que en sujetos desnutridos la caída de la leptina sea uno de los factores por los que aumenta la predisposición a las infecciones.
Leptina y adiponectina son las adipoquinas que más se han estudiado.
La leptina se produce principalmente en los adipocitos maduros, aunque se ha detectado además en el fondo gástrico, en el músculo esquelético, el hígado y la placenta. Se descubrió 1994 por el grupo de Friedman, mediante clonación posicional por mutación de un gen simple en el ratón ob/ob31.
Para Unger, el hecho de que las ratas lipodistróficas se recuperen al trasplantar tejido adiposo o al administrarles leptina muestra que la secreción hormonal de los adipocitos es fundamental para proteger a la célula del daño lipotóxico32.
Es extremadamente interesante la hipótesis de la actividad antiesteatósica de leptina, que regularía en tejidos no adiposos la homeostasis intracelular de los AG y los triglicéridos con el objeto de mantener un aporte apropiado para la función celular y evitar la sobrecarga lipídica. Los AG de cadena larga proveen bloques para las membranas biológicas, es la fuente de componentes de señales celulares y como solo aporta escasa energía, esta llega desde una fuente extracelular como son los adipocitos, que guardan grandes cantidades de lípidos y los exportan según la demanda. Así, las células no adiposas tienen una reserva enorme de energía externa sin que se comprometan sus lípidos intracelulares, que se necesitan para funciones esenciales y elementales33.
Durante la sobrecarga de triglicéridos, los lípidos se atesoran primariamente en los adipocitos y no se distribuyen por igual en todas las células. Por eso es imprescindible que, mientras se depositen triglicéridos, los adipocitos envíen una señal que no permita la acumulación de las grasas en otros tejidos y las confinen en las células adiposas (cuyo contenido de triglicéridos varía en un amplísimo rango, según el ingreso de grasas). Unger consideró que esa señal es la leptina, que produce un monopolio de grasas por los adipocitos de tal manera que el contenido de triglicéridos en tejidos no adiposos está confinado a límites estrechos, sin relación con la ingesta de alimentos, y además tiene varias vías metabólicas, más bien dirigidas a la oxidación de AG que a la lipogénesis34. Así, en periodos de sobrenutrición se disipa la energía innecesaria del exceso de AG como calor y en esta actividad oxidativa tienen una intervención destacada las enzimas desacoplantes35.
La leptina protege ante la oferta en exceso de alimentos, pues carga los triglicéridos en los adipocitos, pero cuando se desarrolla la leptinorresistencia aparece el depósito de grasas en tejidos no adiposos (esteatosis).
La adiponectina es una hormona proteica de molécula monomérica con 244 aminoácidos que se sintetiza exclusivamente en el tejido adiposo (los adipocitos omentales secretan más adiponectina que los subcutáneos), se vincula a la sensibilidad sistémica a la insulina e influye en el metabolismo de la glucosa y de las grasas (uno de los «eslabones» entre lípidos y glúcidos). Se encuentra en altas concentraciones en sangre (500-3.0000 μg/L) y circula en 2 isoformas36.
Los niveles de adiponectina son más elevados en mujeres (¿diferencia en tamaño y distribución de adipocitos?), se relacionan con la insulinosensibilidad (e inversamente con insulinorresistencia), la aterosclerosis y la enfermedad cardiovascular y tiene acción antiinflamatoria (con analogía estructural con TNFα). La adiponectina circulante desciende en la obesidad, la insulinorresistencia y la DMT2, pero más aún si esas condiciones se acompañan de enfermedad cardiovascular37.
No se han identificado claramente ni los receptores de adiponectina ni las vías de señal que median en sus efectos metabólicos. El nexo de la adiponectina como protector de enfermedad cardiovascular sería directo e indirecto, ya que posee efectos sobre el endotelio, en el que suprime la respuesta inflamatoria, inhibe la proliferación celular de músculos lisos vasculares y frena la conversión de macrófagos a células espumosas. En cambio, su descenso acentúa la proliferación neointimal en repuesta a la injuria (por corte vascular) y se vincula al aumento de triglicéridos, de LDL pequeñas densas y de Apo B38.
Los niveles de adiponectina se elevan cuando los obesos adelgazan, lo que se podría justificar por el hecho que los adipocitos plenos de triglicéridos producen menos adiponectina que los pequeños y maduros (a su vez, con mayor insulinosensibilidad). En cultivo de adipocitos, la insulina aumenta la expresión de adiponectina. Se podría postular que la adiponectina es baja en la DMT2 debido a que los adipocitos tienen una pobre sensibilidad a la insulina. Pero para deFronzo la disminución de la adiponectina reflejaría «el síndrome de la célula adiposa disfuncional (adiposopatía)»39.
Yamahuchi, Gavrilova y colaboradores lograron con la combinación de leptina y de adiponectina que se normalizaran los niveles de glucosa en la rata con diabetes lipoatrófica. Observaron que hubo menos depósito de triglicéridos en el hígado y en el músculo, se mejoró la expresión de genes involucrados en el uso y el transporte de lípidos y aumentó la oxidación de las grasas en los miocitos40.
Se ha demostrado que las tiazolidenedionas (agentes insulinosensibilizadores agonistas de PPAR) regulan la expresión de adiponectina en el TAB y en sus niveles plasmáticos, y se ha propuesto que estas formas de regulación constituyen los mecanismos más importantes por el que estos fármacos inducen la mejoría de la insulinorresistencia vinculada a la obesidad y a la DMT2.
El ataque de las grasas: lipotoxicidad y lipoapoptosisUn adipocito con función normal protege contra el daño lipotóxico y tiene actividad antiesteatócica. El término de lipotoxicidad no incluye solo la noxa que resulta de la sobrecarga lipídica por exceso de AG vs. la capacidad de oxidación, sino que se suma y se amplía a las consecuencias del exceso de glucosa («gluco-lipotoxicidad»).
Aún no se ha dilucidado si la acumulación de triglicéridos en células magras es la causa de la lipotoxicidad o simplemente un marcador. Una vez que un AG ingresa en la célula, tiene diferentes posibilidades:
- 1.
oxidarse;
- 2.
guardarse en reserva como triglicérido o
- 3.
dirigirse a vías alternativas no oxidativas con efectos deletéreos, pues inducen la formación de sustancias nocivas, como las ceramidas, o de moléculas reactivas de lípidos que favorecen la disfunción o aceleran la apoptosis celular.
El metabolismo no oxidativo de los productos de los AG de cadena larga provoca el cúmulo de triacilglicerol y ceramidas (es posible incluso en la célula β) y causaría primero lipotoxicidad mediada por óxido nítrico y luego muerte celular o lipoapoptosis. No está aún claramente explicado el mecanismo molecular del inicio de la leptinorresistencia. Esto último se produce a través de la activación de caspasas (cisteín-proteasas, cuyo residuo cisteína media la rotura de otras proteínas y son esenciales en la muerte celular programada).
Las ceramidas pueden, a su vez, aumentar el óxido nítrico que causa la apoptosis en células cargadas de lípidos41. De la misma forma, existen experimentos que muestran que la generación de especies de oxígeno reducido también produce lipotoxicidad por una vía independiente de las ceramidas42.
Las células β y los cardiomiocitos son células propensas a sufrir la disminución de su población celular por acción de los lípidos.
La influencia de las grasas sobre la célula betaLos lípidos muestran efectos sobre la función y la sobrevida de las células β; sin embargo, no se conocen la totalidad de los eventos que conducen a la insulinodeficiencia.
Los AG ejercen un efecto dual sobre la secreción de insulina. Los AGL son imprescindibles para la secreción de insulina en ayunas y actúan en los últimos pasos del proceso de acoplamiento-secreción con acción sobre la exocitosis del gránulo de insulina. También se describen otros mecanismos posibles, entre los que se destaca la unión de los AG con su receptor GPR-40 (recientemente descripto), que tiene una alta expresión en la célula β43.
Los estudios por Sako y Grill, en 1990, fueron los primeros en los que se comparó el efecto en corto y largo plazo de la hiperlipidemia experimental sobre la secreción de insulina. Demostraron que la infusión de lípidos al principio estimula la secreción de insulina inducida por la glucosa, mientras que por un largo plazo inhibe la secreción. Por primera vez explicaron en investigación básica la dependencia del tiempo en la respuesta de la célula β a las grasas44.
La exposición de los islotes a niveles altos de AG mejora la secreción de insulina si el estímulo de glucosa es bajo; en cambio, suprime la secreción de insulina si el estímulo de glucosa es elevado. Se han propuesto diversos modos de acción45-47:
- 1.
la alteración en la expresión de genes que codifican las enzimas del metabolismo de la glucosa y aumentan de los factores que elevan la oxidación de los AG;
- 2.
el aumento del desacoplamiento en las mitocondrias, con lo que desciende el ATP, que es esencial en la secreción de insulina (sobreexpresión de proteína desacoplante 2 [UCP-2]);
- 3.
elevación de las ceramidas y de especies de oxígeno reducido;
- 4.
la inducción de apoptosis y reducción de su capacidad de reproducción, como se ha observado por la acción del ácido palmítico (saturado) in vitro.
El tipo de AG también afecta a las células. Los AG saturados favorecerían la apoptosis betacelular (vía PKC-delta); en cambio, los AG insaturados protegerían contra la actividad apoptótica de los saturados.
El contenido graso en el páncreas se relaciona en forma negativa con la secreción de insulina; sin embargo, la dificultad para medir los triglicéridos en la célula β no permite que se defina con claridad aún la totalidad de las funciones que cumplen en el tejido pancreático. Así también se especula que la acción adversa de los niveles de AGL sobre la respuesta secretoria de insulina, acentúa el efecto glucotóxico de la hiperglucemia sobre la célula β (glucolipotoxicidad) y aceleraría la evolución de la enfermedad.
Numerosas y complejas nuevas líneas de investigaciónSe reconoce que existen factores de transcripción que son clave para la regulación del metabolismo y la homeostasis de los lípidos, y tal vez influyen en el proceso de lipotoxicidad: la familia de la proteína-1 ligadora de elementos reguladores de esteroles (SREBP1 o sterol regulatory element-binding protein 1), ya que controlan la expresión de un número de enzimas que se requieren para la síntesis endógena de colesterol, AG, triglicéridos y fosfolípidos. A su vez, los esteroles/AG/triglicéridos constituyen un feed-back negativo que inhibe la activación de SREBP para que se reduzca la síntesis de lípidos.
Hay 3 isoformas con diferentes efectos: SREBP-1a, SREBP-1c y SREBP-2. Estudios experimentales han mostrado que la SREBP1c es una isoforma que se regula por nutrientes y modula por la insulina, que también se expresa en tejido TAB y se vincula con la síntesis de novo de AG y triglicéridos48. Se especula con la posibilidad de que la ausencia de SREBP1c en los adipocitos afecte al almacenamiento de triglicéridos, lo que aumentaría el flujo de AGL en circulación y hacia otros tejidos. En definitiva, provocaría lipotoxicidad48.
Otro punto en desarrollo son las investigaciones sobre la familia de PPAR, íntimamente involucrada con el metabolismo de lípidos, hidratos de carbono e incluso se describen 2 subtipos de estos receptores nucleares (que representan el sitio de acción de agonistas sintéticos): PPAR-α y PPAR-γ. Al subtipo alfa se lo considera en relación con el proceso oxidativo de los AG, mientras que al gamma se lo vincula con la redistribución grasa (de visceral a subcutánea) y con los niveles de adiponectina, una adipoquina a la que aumenta. Al activar el PPAR-γ hay un descenso de los AGL plasmáticos y la mejoría de la sensibilidad a la insulina. El efecto de los agonistas de PPAR-α sobre el metabolismo de lípidos podría también producir cierta mejora en la sensibilidad a la insulina. Se dispone de agonistas que activan selectivamente PPAR-α (p. ej., fibratos) o PPAR-γ (p. ej., tiazolidinedionas), aunque también existen agonistas duales PPAR-α/γ que suponen más amplios beneficios que aquellos que son agonistas específicos.
El concepto sobre la lipogénesis de novo ha tenido vaivenes y se ha subestimado su valor y posible mecanismo de protección de la acumulación exagerada de grasas, ya que la conversión de glúcidos en grasas es un proceso que requiere de alta energía, si se compara con el depósito de lípidos exógenos como grasa corporal. Para guardar grasas, se utiliza alrededor del 25% de la energía que contienen los hidratos de carbono; en cambio, solo se necesita un 2% del gasto de los triglicéridos dietarios para depositarse. Sin embargo, aún existen numerosos interrogantes y controversias con referencia a la lipogénesis de novo, que incluyen su importancia fisiológica, su influencia en procesos patológicos o incluso sobre el lugar principal de producción (¿hígado y/o tejido adiposo?)49,21.
Energía y defensa son elementales para la vida, por eso los sistemas metabólico e inmunológico, aunque son independientes, tienen funciones que se interrelacionan y se regulan uno a otro. Muchas hormonas, citoquinas, proteínas de señal, factores de transcripción y lípidos bioactivos pueden cumplir funciones tanto metabólicas como inmunológicas. Se reconoce la unión entre desnutrición e inmunodeficiencia, pero hoy se alerta sobre el vínculo de la obesidad, la diabetes, la enfermedad grasa del hígado y la aterosclerosis con la respuesta inflamatoria crónica por una anormal producción de citoquinas, el aumento de reactantes de fase aguda y la activación de vías de señal inflamatoria. Se especula que la respuesta inflamatoria leve y amplia que se observa en las enfermedades metabólicas se inicia en los propios adipocitos disfuncionales o potencialmente en sus estructura vecinas perturbadas por el crecimiento adipocitario, como son las células reticuloendoteliales y los precursores adipocitarios. También hay macrófagos que infiltran al tejido adiposo en expansión y es probable que estas células produzcan mediadores inflamatorios por sí mismos o por coacción con los adipocitos. Algunos de estos mediadores exhiben patrones de expresión o influyen en la actividad de la insulina.
ConclusionesEntre las misiones y las funciones de los glúcidos y de las grasas no existe solo relación recíproca, sino también dependencia metabólica. La elevación de la glucemia en el periodo prandial (absorción) se acompaña de la secreción de insulina que suprime la liberación de AG en los adipocitos, y se estimula la captación y la utilización de la glucosa por el músculo. En los periodos interprandiales (postabsorción), caen la glucosa y la insulina, y desde el tejido adiposo se liberan AGL, que constituyen el combustible principal del músculo.
El adipocito, el tejido adiposo y las grasas circulantes cumplen un papel trascendente en la maquinaria metabólica y es notable cómo la disfunción adipocitaria influye sobre el estado del resto de los órganos, principalmente los del «triunvirato de deFronzo: músculo, hígado y célula beta». Hoy se extiende en forma directa o indirecta (pasando por enfermedades metabólicas) al aparato cardiovascular.
Los conceptos del ciclo glucosa-AG (Randle) y la hipótesis portal de la insulinorresistencia constituyeron los intentos tradicionales de explicación racional que se utilizaron durante años para describir el vínculo entre los metabolismos glucídico y lipídico, la obesidad, el fenómeno de resistencia a la insulina y la diabetes.
Investigaciones posteriores evidenciaron que para evitar la producción de alteraciones metabólica el adipocito tiene un cometido esencial: mantener la capacidad para acumular y oxidar lípidos, y de alguna manera, proteger al resto de los tejidos. Cuando pierde dichas cualidades, los AG circulan en mayor cantidad e incluso forman derivados no oxidativos que se acumulan dentro de las células y alteran la función (lipotoxicidad) o incluso aceleran la apoptosis en órganos esenciales para la homeostasis metabólica. Esto se conoce como el paradigma de la ectopia grasa.
Este concepto se sostiene por 3 líneas de evidencia50:
- 1.
El aumento del volumen de la célula adiposa. El gran tamaño adipocitario representaría el fallo de la masa de tejido adiposo para expandirse y así ajustar cualquier aumento del flujo de energía. En las lipoatrofias con insulinorresistencia severa y diabetes se reconoce que el cuadro es consecuencia del depósito ectópico de lípidos en el hígado, el músculo y la célula β.
- 2.
El envío y el asiento de lípidos en el músculo esquelético, el hígado y, probablemente, la célulaβ, como sucede en los pacientes más obesos. Así, diversos estudios muestran que existe una alta correlación entre la infiltración lipídica muscular y hepática y el grado de resistencia a la insulina.
- 3.
Incapacidad para oxidar la totalidad de las grasas disponibles, ya sea en el propio adipocito como en los tejidos magros, que no se desembarazan y acumulan grasas en sus estructuras.
De allí que algunos autores interpretaran la insulinorresistencia por las grasas como una «lipodistrofia adquirida». El aumento de los lípidos intramiocelulares provoca defectos de señal, reduce el transporte de glucosa que depende de la insulina en el músculo, disminuye la síntesis de glucógeno y altera la supresión de la producción endógena de glucosa por el hígado.
Una prueba clínica de común observación es el asiento de las grasas en el hígado. Se traduce en el denominado hígado graso, de sorprendente prevalencia y no muy severas consecuencias para la mayor parte de los pacientes, aunque se reconoce que un grupo pequeño puede evolucionar a esteatohepatitis y finalizar con una cirrosis de tipo no alcohólica. Es el órgano central del metabolismo, aunque aún no se conoce en profundidad los mecanismos que producen resistencia hepática a la insulina, como consecuencia de las grasas.
Desde la fisiopatología se considera que existe un patrón adipo-insular. Su funcionalidad mantiene una íntima relación con la del eje hipotálamo-hipofiso-corticoadrenal, que involucraría 2 señales endógenas (insulina y glucocorticoide, respectivamente) con capacidad adipogénica por excelencia. La disfunción de una o ambas estructuras (insulinorresistencia y/o hipercortisolismo) facilitaría la expansión hipertrófica de la masa de TAB (fundamentalmente el omental perivisceral), caracterizada por disfunción endocrino-metabólica del adipocito blanco, con su impacto sobre el desarrollo de la obesidad y la DMT2. El «paradigma endocrino» por el que el tejido adiposo secreta una variedad de señales con un potente efecto metabólico sobre otros tejidos, cercanos o distantes51, y que tendrían fundamental influencia sobre los mecanismos productores de insulinorresistencia (leptina, adiponectina, TNF-α, resistina, etc.) y en el estado inmunológico (inflamación de grado leve), endocrino y vascular.
Se investigan con entusiasmo las causas y las consecuencias del cúmulo de grasa perivascular, pericardial y miocárdica. Perseghin dedicó gran parte de su conferencia, y premio Camilo Golgi, en el congreso de la Asociación Europea para el Estudio de la Diabetes 2009, a mostrar la evolución de estos conocimientos y posibles vínculos con la aterosclerosis y la insuficiencia cardiaca. El contenido elevado de lípidos miocárdicos en los obesos y diabéticos se vincula con la disfunción mitocondrial y contráctil, y podría preceder al desarrollo de insuficiencia cardiaca. En una reciente publicación se concluyó que la grasa pericardial se asoció con mayor fuerza al desarrollo de enfermedad cardiaca coronaria que la medición simple antropométrica de la obesidad abdominal52.
Todo esto en el devenir de nuevos aspectos sobre la disminución de la capacidad oxidativa en la cadena respiratoria mitocondrial, el renacer del concepto de lipogénesis de novo, que favorecerían el atesoramiento de grasas intracelular (uno por fallos en la eliminación y otros por producción de grasas desde carbohidratos y proteínas). Unger y McGarry argumentaron que en tejidos magros existen pequeñas reservas intracelulares de grasas que mantienen una severa regulación de funciones esenciales, pero si aparece una sobrecarga de lípidos el fenómeno conduciría a disfunción celular (lipotoxicidad) y a la muerte celular (lipoapoptosis).
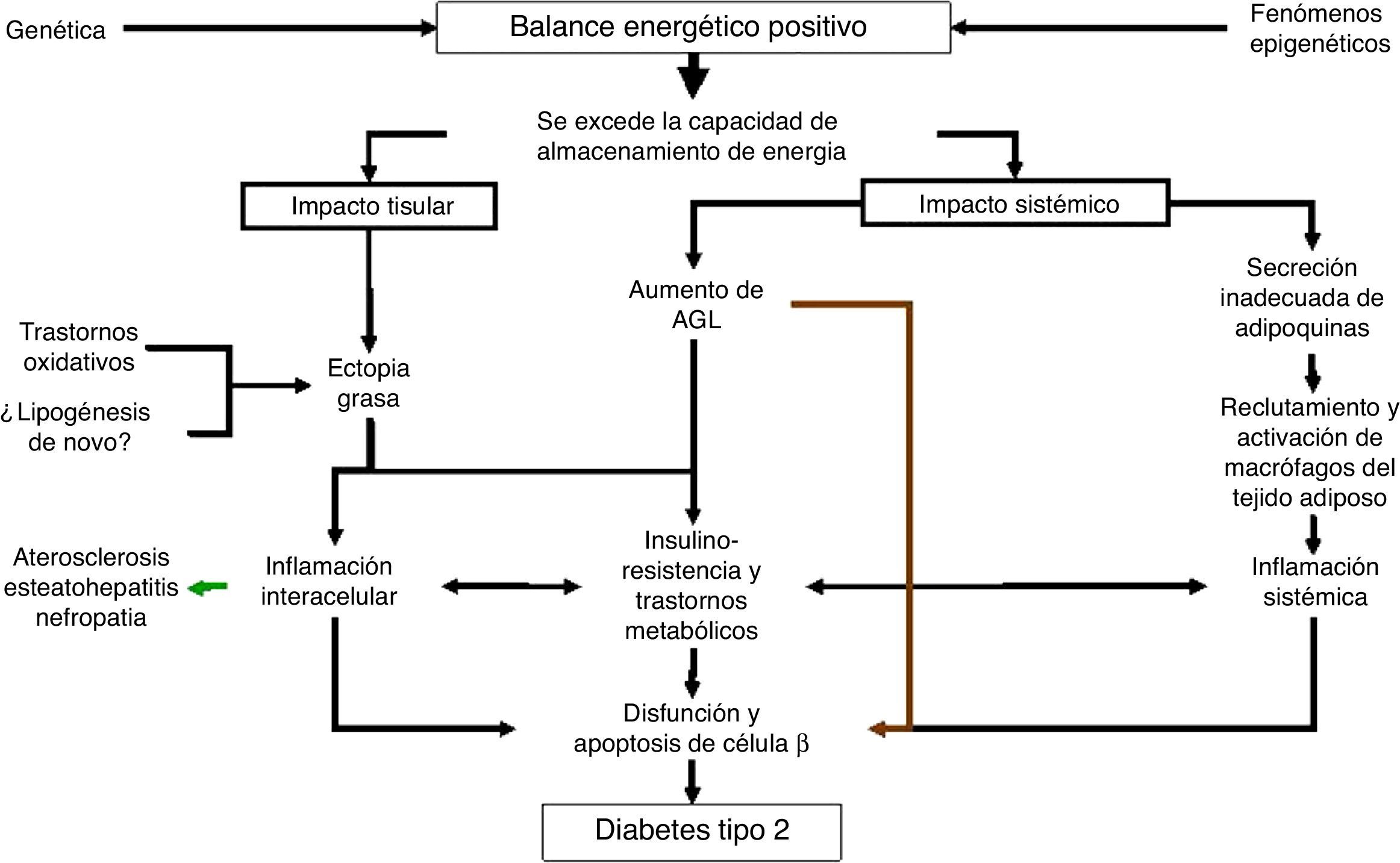
Es una decisión audaz. Sin embargo, bajo la luz de los conocimientos actuales, se podrían integrar las hipótesis y las evidencias y deducir que no hay una única causa, recorrido ni mecanismo (fig. 5). En un trasfondo génico particular y fenómenos epigenéticos que desencadenan o asisten, se excede la capacidad para almacenar energía por parte del tejido adiposo. Se desencadenan así 2 tipos de fenómenos, uno tisular y otro sistémico. El primero se representa por la ectopia grasa en tejidos magros, tal vez acentuada por trastornos en la maquinaria oxidativa y la posibilidad de formación de grasas desde fuentes no lipídicas. Desde el camino sistémico se reconoce tradicionalmente el aumento de los AGL y la producción de adipoquinas por parte de los adipocitos, entre ellas leptina y adiponectina, cuya anomalía influye causalmente en el asiento ectópico de grasas. Es claro hoy que los AGL contribuyen en forma decisiva en el fenómeno de insulinorresistencia que provoca un esfuerzo funcional agotador a la célula beta, pero también en cantidades excesivas y en exposiciones prolongadas, en forma directa la intoxica. Asimismo, las adipoquinas son moléculas de señal que influyen sobre la sensibilidad a la insulina, pero también sobre fenómenos inflamatorios que terminarán lesionando gravemente la célula beta (y otros órganos, como el árbol arterial, el hígado y el riñón).

Además, hay que integrar el tejido adiposo calorigénico, no solo por la novedad que significa la presencia de células beige en el humano con funciones de disipación de calor y la hormona irisina, que abrió un nuevo panorama para interpretar el gasto energético al constituir una mioquina, que vincula la actividad muscular y la pardización del TAB.
Finalmente, la tormentosa relación entre el tejido adiposo y el islote de Langerhans va más allá del esfuerzo funcional que impone la insulinorresistencia periférica a la célula β y tiene sustento en los efectos directos de los lípidos o sus derivados sobre la biosíntesis y la secreción de insulina. Y sin déficit de insulina, no hay diabetes.
Es una historia larga de encuentros y desencuentros, desde las moscas de Minkowski a las demostraciones de Krebs, Randle, McGarry, Reaven, deFronzo, Ferraninni, Shulman, Unger y Scherer como puntos notables que alientan y despiertan entusiasmo por un tema que, en la práctica, se traduce en una epidemia, que aún no muestra signos de mejora clínica (ni epidemiológica).
A estos se suman nuevos aspectos que, lejos de permitir una explicación consolidada de los mecanismos íntimos de la insulinorresistencia, le agregan una apasionante cuota de incertidumbre al rompecabezas que resulta tratar de explicar uno de los fenómenos metabólicos más trascendentes, por su influencia directa sobre la salud de las personas.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónEl estudio fue financiado por el Proyecto FPREDM-052015 (Fondation pour la Recherche en Endocrinologie, Diabetologie et Metabolisme, Lausanne, Suisse).
Conflicto de interesesLos autores declaran que no existe conflicto de interés alguno y que ellos son los responsables del contenido y de la escritura del manuscrito.