


El déficit congénito de surfactante es una entidad de diagnóstico inhabitual en recién nacidos. Se reporta un caso clínico de déficit de proteína B del surfactante, se revisa el estudio, tratamiento y diagnóstico diferencial de los déficit de proteínas del surfactante y enfermedad crónica intersticial de la infancia.
Caso clínicoRecién nacido de término que cursa dificultad respiratoria, con velamiento pulmonar recurrente y respuesta transitoria a administración de surfactante. El estudio inmunohistoquímico y genético confirmaron diagnóstico de déficit de proteína B de surfactante.
ConclusionesLa enfermedad pulmonar congénita requiere un alto índice de sospecha. El déficit de proteína B de surfactante genera un cuadro clínico progresivo y mortal en la mayoría de los casos, al igual que el déficit de transportador ATP binding cassette, sub-family A member 3 (ABCA3). El déficit de proteína C es insidioso y puede presentarse con un patrón radiológico pulmonar intersticial. Debido a la similitud en el patrón histológico, el estudio genético permite una mayor certeza en el pronóstico y la posibilidad de entregar un adecuado consejo genético.
Congenital surfactant deficiency is a condition infrequently diagnosed in newborns. A clinical case is presented of surfactant protein B deficiency. A review is performed on the study, treatment and differential diagnosis of surfactant protein deficiencies and infant chronic interstitial lung disease.
Case reportThe case is presented of a term newborn that developed respiratory distress, recurrent pulmonary opacification, and a transient response to the administration of surfactant. Immunohistochemical and genetic studies confirmed the diagnosis of surfactant protein B deficiency.
ConclusionsPulmonary congenital anomalies require a high index of suspicion. Surfactant protein B deficiency is clinically progressive and fatal in the majority of the cases, similar to that of ATP binding cassette subfamily A member 3 (ABCA3) deficiency. Protein C deficiency is insidious and may present with a radiological pulmonary interstitial pattern. Due to the similarity in the histological pattern, genetic studies help to achieve greater certainty in the prognosis and the possibility of providing adequate genetic counselling.
El surfactante pulmonar es un complejo molecular localizado en la interfase aire-líquido del pulmón. Está compuesto por lípidos, en su mayoría fosfolípidos, y en un 2–3% por proteínas específicas denominadas proteínas A, B, C y D («surfactant protein» SP-A, SP-B, SP-C y SP-D). SP-B y SP-C son proteínas hidrofóbicas, sintetizadas por neumocitos tipo ii, que disminuyen la tensión superficial y el colapso alveolar. La ATP binding cassette, sub-family A, member 3 (ABCA3) es una proteína encargada del transporte de fosfolípidos y proteínas del surfactante transmembrana desde el citosol hacia los cuerpos lamelares, organelos intracelulares encargados de almacenar lípidos y proteínas del surfactante1, con control génico independiente de la producción de proteínas.
El déficit congénito de surfactante es una enfermedad respiratoria muy poco frecuente, que no se considera dentro del diagnóstico diferencial inicial del síndrome de distrés respiratorio del recién nacido. La evolución respiratoria es muy variable, dependiendo de la proteína afectada. Esta puede presentarse como un cuadro respiratorio grave, progresivo, con una elevada mortalidad a corto plazo; o puede mostrar un inicio insidioso, lentamente progresivo. El objetivo es mostrar un caso clínico y revisar el estudio diagnóstico y el eventual tratamiento.
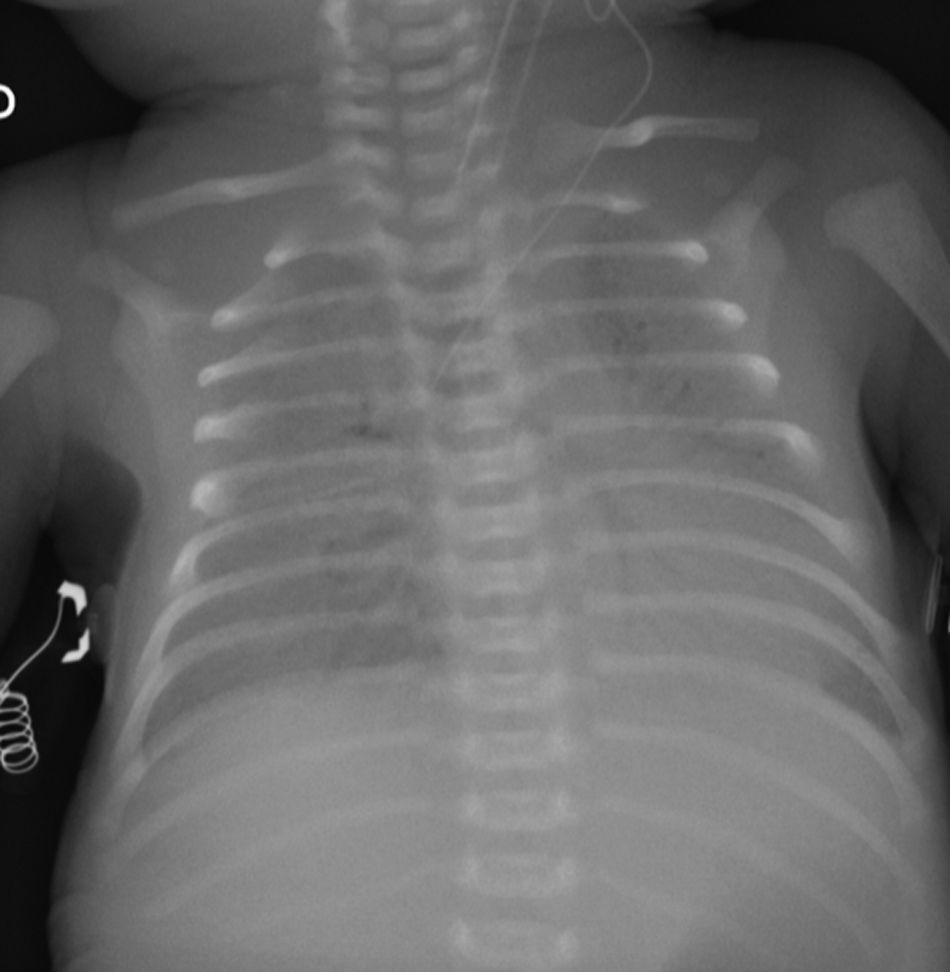
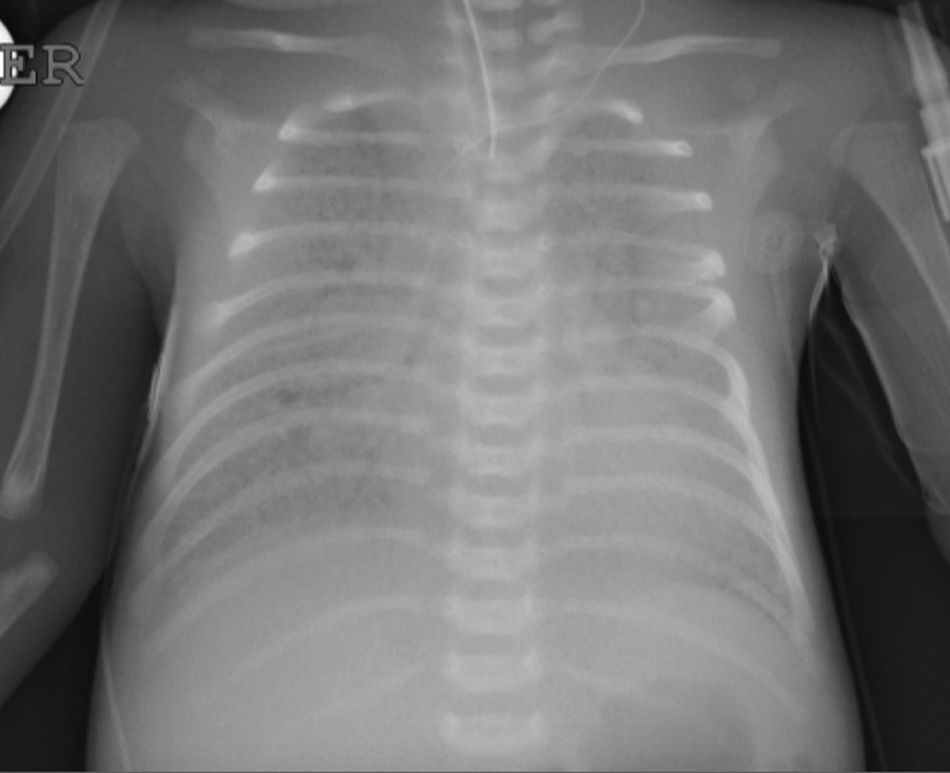
Caso clínicoRecién nacido de término de 37 semanas, pequeño para la edad gestacional. Padres consanguíneos, sin antecedentes mórbidos personales. Madre primigesta, cesárea electiva a las 37 semanas. Nació con APGAR 8-9. Rápidamente evolucionó con dificultad respiratoria; radiografía de tórax con patrón con vidrio esmerilado y broncograma aéreo a las 2h de vida. Se administró una dosis de surfactante, con buena respuesta inicial. Deterioro respiratorio a las 12 y 24 h, que requirió 3 dosis de surfactante. Se trasladó a centro con ventilación de alta frecuencia oscilatoria (VAFO) y óxido nítrico (ONi). Ingresó a las 30 h de vida con parámetros ventilatorios altos 30/5 FiO2 100% para saturar 84%. Se conectó a VAFO y recibióe surfactante y ONi. Radiografía de tórax con imagen de vidrio esmerilado desde su ingreso. A los 7 días de vida se controlaron las imágenes (fig. 1) y se inició sildenafil. A los 18 días de vida se logró suspender ONi. Sin elementos de infección bacteriana, aunque se cubre con antibióticos, se descartó citomegalovirus y Pneumocystis jirovesi. Recibió corticoides sin mejoría clínica ni posibilidad de retiro de VAFO. Dada la evolución clínica con imágenes persistentes de membrana hialina (fig. 2), consanguinidad, se planteó déficit de proteína B del surfactante y se realizó biopsia pulmonar a los 21 días. Histología compatible con déficit de surfactante, con fibrosis e infiltrado mononuclear con epitelio alveolar constituido principalmente por neumocitos tipo ii sin membranas. La histoquímica confirmó déficit de proteína B del surfactante. No se realizó microscopia electrónica. Se envió muestra de sangre en papel filtro a la Universidad de Washington, confirmándose homocigoto para nueva mutación de déficit de SP-B. Se presentó a Comité de Ética, considerando un diagnóstico certero de mal pronóstico y sin posibilidad terapéutica, se consultó a especialistas de centros internacionales acreditados. Presentó deterioro respiratorio progresivo y falleció a los 56 días. Se envió a los padres a consejo genético.


Si bien en la literatura el déficit de surfactante se describe como poco frecuente, en la medida en que es estudiado ha aumentado su prevalencia. Incluso existe un reporte previo en nuestro país2. Este debe sospecharse en recién nacidos con una presentación clínica de enfermedad de membrana hialina que recurre en el tiempo a pesar del aporte de surfactante, y en cuadros respiratorios crónicamente progresivos en que se descartan causas infecciosas habituales y ciertas enfermedades pulmonares.
Descartados displasia broncopulmonar, fibrosis quística, infecciones recurrentes, cardiopatía con obstrucción del retorno venoso, frente a un cuadro respiratorio crónico debe considerarse el grupo de enfermedades definidas como enfermedad intersticial crónica de la infancia (CHILD de children's interstitial lung disease), que incluye anomalías del surfactante, hiperplasia celular neuroendocrina de la infancia, displasia alvéolo capilar asociada con malalineamiento de venas pulmonares y enfermedad pulmonar intersticial por glucogenosis, entre otros3. En el recién nacido o lactante menor de inicio neonatal con enfermedad crónica debe estudiarse la enfermedad genética de síntesis y transporte de proteínas de surfactante3,4.
El análisis inmunohistoquímico de SP-B y el estudio genético permitieron realizar un diagnóstico certero. El enfrentamiento clínico difiere por el conocimiento etiológico de la enfermedad respiratoria y por el tipo de proteína afectada, ya que la ausencia de SP-B es letal. Desde el punto de vista ético-legal, es una situación muy compleja, sobre todo para la familia, que observa un niño bien formado y con reactividad normal, por lo que es muy difícil aceptar la falta de alternativa terapéutica actual.
El déficit de SP-B es el más frecuente. Sin embargo, existe también el déficit de SP-C, ABCA3 y factor de transcripción tiroideo, cada uno con diferente presentación clínica y pronóstico5. Para el diagnóstico de esta enfermedad puede realizarse tomografía axial computada, biopsia pulmonar y estudio genético5.
La mayoría de los déficit de SP-B se heredan en forma autosómica recesiva4,5. La incidencia es de 1/1.000 individuos6. La SP-B está localizada en el cromosoma 2, la mutación más frecuente es 121ins2, que resulta en la ausencia de proteínas proSP-B y SP-B4,5,7. Evolucionan desde el nacimiento con distrés respiratorio grave y progresivo. En la mayoría resulta rápidamente fatal en días o meses, sobre todo si la expresión del déficit de SP-B es completa4,7. La frecuencia de alelos, genotipos y polimorfismos puede variar entre diferentes grupos étnicos. Su estudio puede ayudar a comprender el riesgo individual (y étnico) de presentar déficit congénito de surfactante8.
El déficit de SP-C está relacionado con distrés respiratorio en el recién nacido y con enfermedad pulmonar intersticial en niños mayores9. Los síntomas respiratorios asociados a mutaciones del SP-C usualmente comienzan los primeros meses de vida. Mutaciones del SP-C han sido reportadas no solamente en recién nacidos con distrés respiratorio agudo grave, sino también en niños, e incluso adultos con enfermedad pulmonar intersticial. La mutación más prevalente es I73T (c.218 T>C)9. La presencia de tos, polipnea y decaimiento asociada a un patrón en vidrio esmerilado bilateral en TAC de pulmón apoya la realización de lavado broncoalveolar y estudio genético para SP-C. El pronóstico a largo plazo es variable; puede incluso suspenderse el aporte de oxígeno suplementario9.
Por otra parte, los genes que codifican para ABCA3 son críticos en la formación de cuerpos lamelares en los neumocitos tipo ii5,10. El gen ABCA3 codifica para una proteína de alta expresión a nivel pulmonar que está involucrada en el transporte de fosfolípidos de los cuerpos lamelares11. El déficit de ABCA3 produce una acumulación de lípidos, proteínas y macrófagos en el espacio alveolar con engrosamiento epitelial. En algunos casos, la variante ABCA3 puede resultar en enfermedad respiratoria de curso más insidioso11. Sin embargo, generalmente, está asociado a una alteración severa y altamente letal de la producción de surfactante del neumocito tipo ii10,11. En los casos severos se ha reportado en asociación a alteraciones del procesamiento de SP-B y SP-C12,13.
Respecto al diagnóstico, la biopsia pulmonar lo orienta, aunque tiene limitaciones dado que existe similitud en el patrón histológico de varias entidades con igual presentación clínica. En la deficiencia de ABCA3, de SP-C y del factor de transcripción tiroideo los hallazgos histopatológicos pulmonares primarios son inespecíficos y están caracterizados por hiperplasia celular alveolar de neumocitos tipo ii, engrosamiento del intersticio y fibrosis10. La inmunohistoquímica para SP-B y el análisis ultraestructural de los cuerpos lamelares tienen un rol para determinar el déficit de SP-B y ABCA-3, respectivamente1.
La manera más específica para obtener un diagnóstico etiológico es el estudio genético de mutaciones. El consejo genético es importante, ya que las mutaciones de SP-C y del factor de transcripción tiroideo son dominantes y pueden aparecer espontáneamente10. El estudio genético antenatal de vellosidades coriales y líquido amniótico solo está disponible en laboratorios de investigación14.
El manejo médico es limitado, debido al déficit intrínseco de surfactante. El tratamiento con surfactante exógeno puede o no resultar en una mejoría clínica transitoria. Se ha reportado la futilidad de la administración de hasta 80 dosis de surfactante en la deficiencia SP-B14. El uso de hidroxicloroquina y corticoides en déficit de SP-C es controversial15. El trasplante de pulmón es, hasta el momento, la única alternativa terapéutica cuando la falla respiratoria es irreversible y progresiva. La sobrevida a 5 años en recién nacidos con déficit de SP-B, SP-C y ABCA3 en Estados Unidos es alrededor de 50%13,16. El cuidado posterior al trasplante pulmonar requiere de inmunosupresión a largo plazo y un seguimiento cercano16. La mortalidad es principalmente secundaria a síndrome linfoproliferativo, a infecciones y a bronquiolitis obliterante, todas atribuidas al trasplante de pulmón14. La terapia génica se encuentra en investigación14. El desafío actual es efectuar un diagnóstico antenatal para lograr el traslado de estos pacientes a centros especializados antes del nacimiento16. Localmente es importante buscar la enfermedad crónica intersticial en aquellos pacientes de evolución atípica para los cuadros neonatales más conocidos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos al Dr. Aaron Hamvas, de la Washington University y del St. Louis Children's Hospital por realizar el estudio genético del paciente.
