



El síndrome H es una enfermedad genética extremadamente rara de compromiso multisistémico, el cual clínicamente puede ser reconocido de forma precoz, ofreciendo de manera oportuna un seguimiento, tratamiento específico y asesoramiento genético.
ObjetivoPresentar un caso con características «típicas del síndrome H» para favorecer su identificación precoz.
Caso clínicoVarón de 8 años de edad, evaluado por tumoraciones testiculares, lesiones dérmicas tipo hiperpigmentación con hipertricosis, retraso del lenguaje, talla baja, deformidades articulares, hipoacusia neurosensorial bilateral, anemia, hipergammaglobulinemia y alteraciones óseas. En los estudios histológicos de la piel y las masas testiculares se observó infiltración linfoplasmocitaria. El secuenciamiento del gen SLC29A3 detectó una mutación homocigota c.1087 C>T (p.Arg363Trp; rs387907067) concluyente con el síndrome H, la cual ha sido reportada previamente.
ConclusionesEste es el primer caso reportado en Latinoamérica del síndrome H, cuyas características descritas son parte del espectro clínico. El hallazgo clínico principal, que orienta al diagnóstico, es la hiperpigmentación acompañada de hipertricosis.
H Syndrome is an extremely rare genetic disease, with a multisystemic character and which can be identified in early childhood, offering the opportunity of specific treatment and genetic counselling.
ObjectiveTo present a clinical case with “typical” characteristics of H Syndrome.
Clinical caseThe case is presented of an 8-year-old male patient who presented with testicular tumours and skin lesions characterised by hyperpigmentation with hypertrichosis, language delay, short stature, and joint deformities. He also presented with bilateral sensorineural hearing loss, anaemia, hypergammaglobulinaemia, and bone disorders. Histopathology studies of the skin and testicular masses reported lymphoplasmacytic infiltration. Sequencing analysis of gene SLC29A3 showed the homozygote mutation c.1087 C>T (p.Arg363Trp; rs387907067).
ConclusionsThese findings are consistent with H syndrome, and this is the first reported case in Latin America. The key to the diagnosis is the finding of hyperpigmentation with hypertrichosis.
El síndrome H (MIM 602782) es una enfermedad genética extremadamente rara de compromiso multisistémico. Sus principales características son hipoacusia, hiperglucemia, anomalías cardíacas, hipertricosis, hepatomegalia e hipogonadismo1,2. El rasgo distintivo de la enfermedad es la hiperpigmentación cutánea acompañada de induración e hipertricosis, que inicialmente aparece en la región interna de los muslos y en las crestas tibiales, pero a veces puede ser generalizada1.
Histológicamente en las lesiones se observa un denso infiltrado dérmico y subcutáneo, compuesto principalmente por los histiocitos CD681, posteriormente sustituido por fibrosis3. Además, una característica común es la emperipolesis (presencia de neutrófilos y linfocitos en el citoplasma de otras células)4.
En el año 2008 se implicó el gen SLC29A3 como responsable de la genodermatosis autosómica recesiva denominada síndrome H2. El gen solute carrier family 29 (nucleoside transporter), member 3 codifica una proteína transportadora de nucleósidos (ENT3) y análogos, localizada en los lisosomas y la membrana mitocondrial interna3,5,6.
Las mutaciones del gen SLC29A3 producen la enfermedad conocida en Mendelian Inheritance in Man como síndrome de histiocitosis-linfadenopatía plus, el cual incluye dentro de su espectro fenotípico el síndrome H, así como la histiocitosis Faisabad, enfermedad Rosai-Dorfman y disostoesclerosis, ente otras7. El gen SLC29A3 parece estar implicado en un gran espectro fenotípico, por lo que se debe impulsar a los médicos a estudiarlo, para la detección incluso en cuadros clínicos leves8.
Los síntomas y signos mencionadas son características «típicas» del síndrome H, lo cual hace que sea fácilmente identificable, teniendo como intención principal el diagnóstico precoz y el poder ofrecer un seguimiento y tratamiento más oportuno, así como entregar un asesoramiento genético exacto, considerando que el riesgo de recurrencia es del 25%.
Nuestro objetivo es presentar un caso con características «típicas» del síndrome H, para favorecer su identificación precoz, y ofrecer un seguimiento y tratamiento más oportuno, así como entregar un asesoramiento genético.
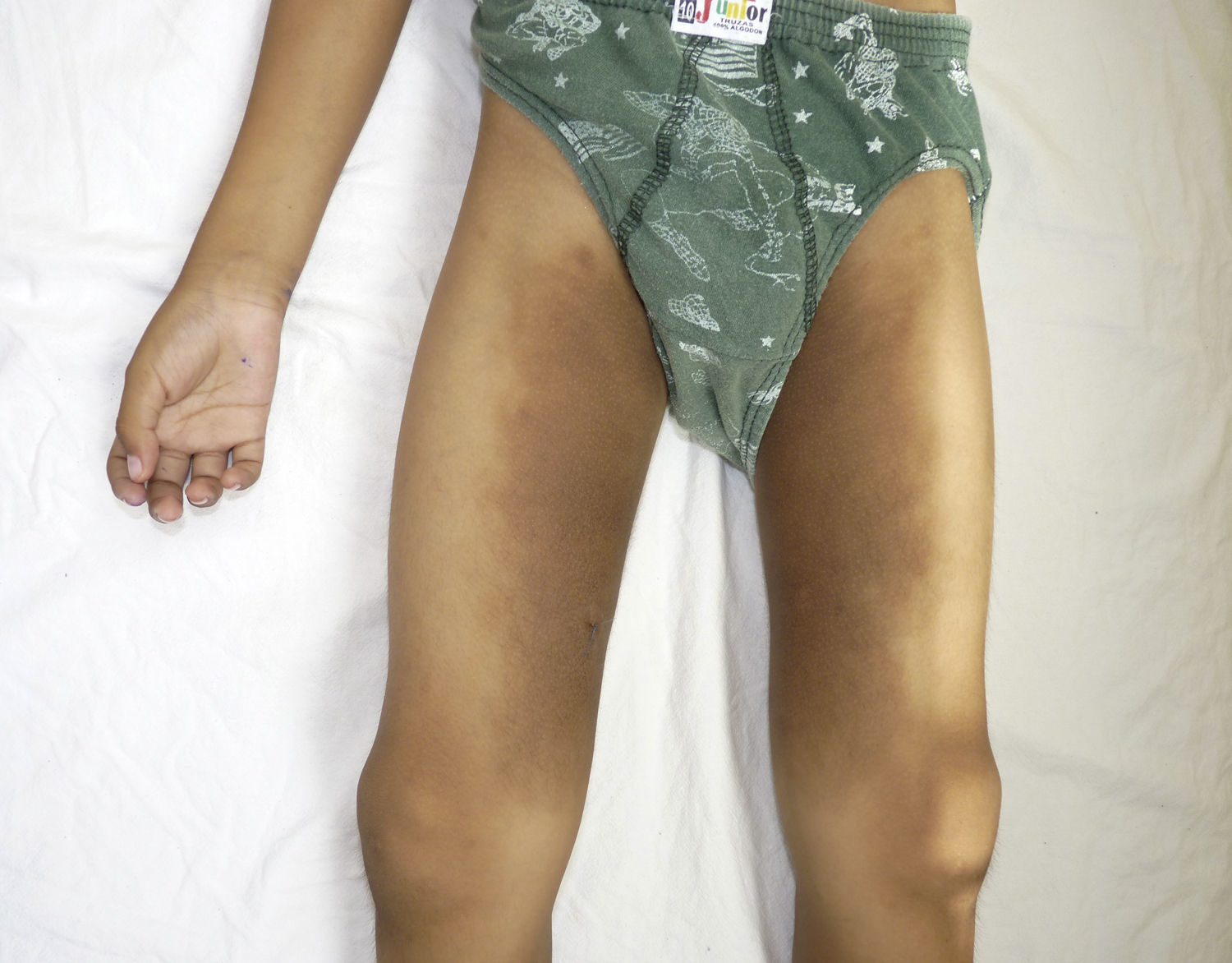
Caso clínicoPaciente varón de 8 años de edad, quien desde los 5 años presentó tumoraciones indoloras en ambos testículos. A los 7 años aparecieron lesiones hiperpigmentadas en incremento del tamaño al nivel de los muslos (fig. 1), asociadas a talla baja, retraso del lenguaje, aumento de volumen de las articulaciones, anemia e hipoacusia neurosensorial bilateral. Sin antecedentes prenatales de importancia, nacido de parto vaginal, domiciliario, con un peso de 2.000g. Con historia de retraso del desarrollo psicomotor.
 que afecta casi la totalidad de la región medial del muslo y que se extiende hacia la región anterior (bilateral).")
Ambos padres procedìan de caseríos aledaños del Cerro Mocho, capital del distrito de Escudero, Provincia de Sullana, Región de Piura, tuvieron un hijo mayor fallecido de cardiopatía congénita no especificada a los 2 meses de edad, sin antecedentes de consanguinidad.
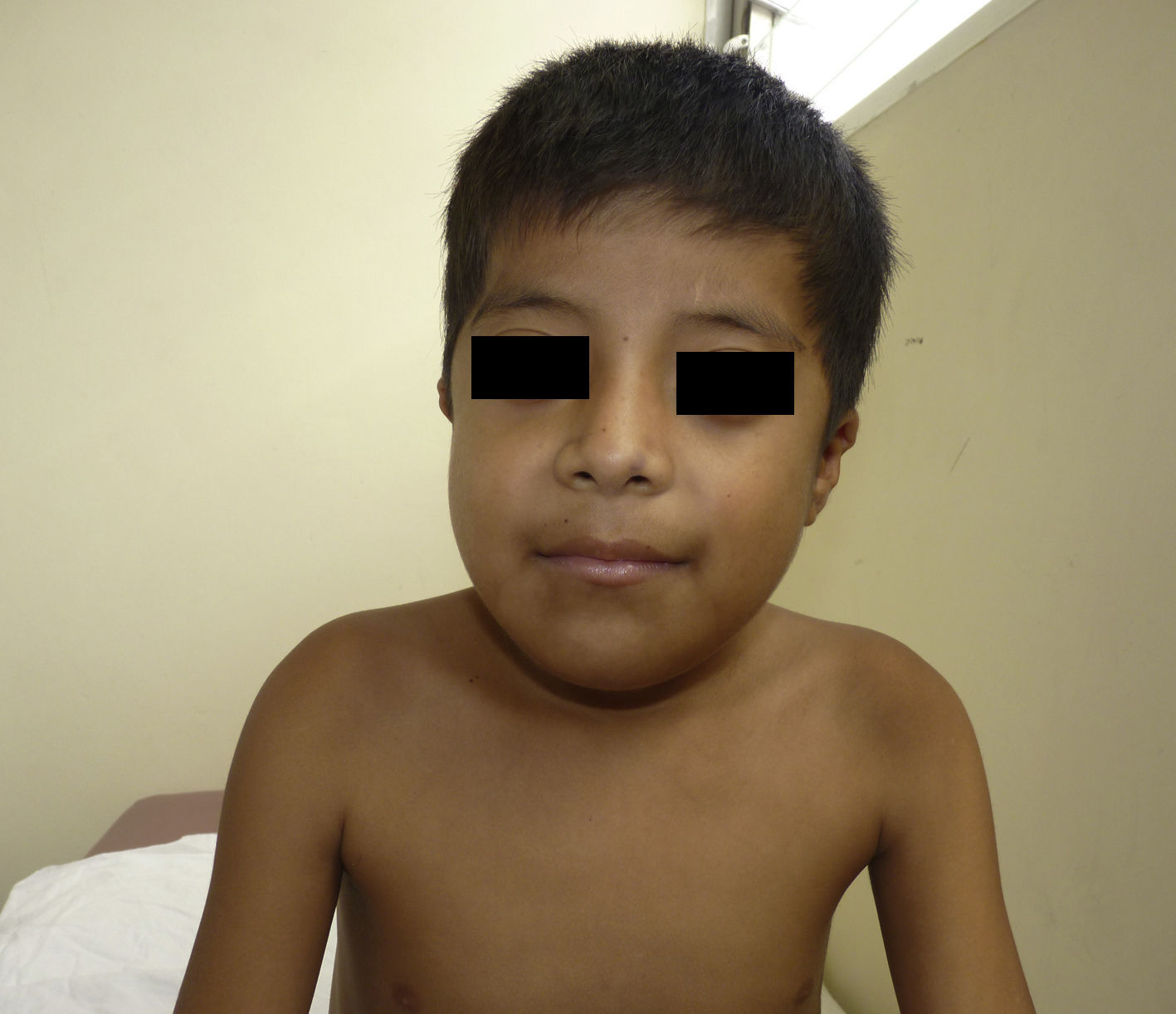
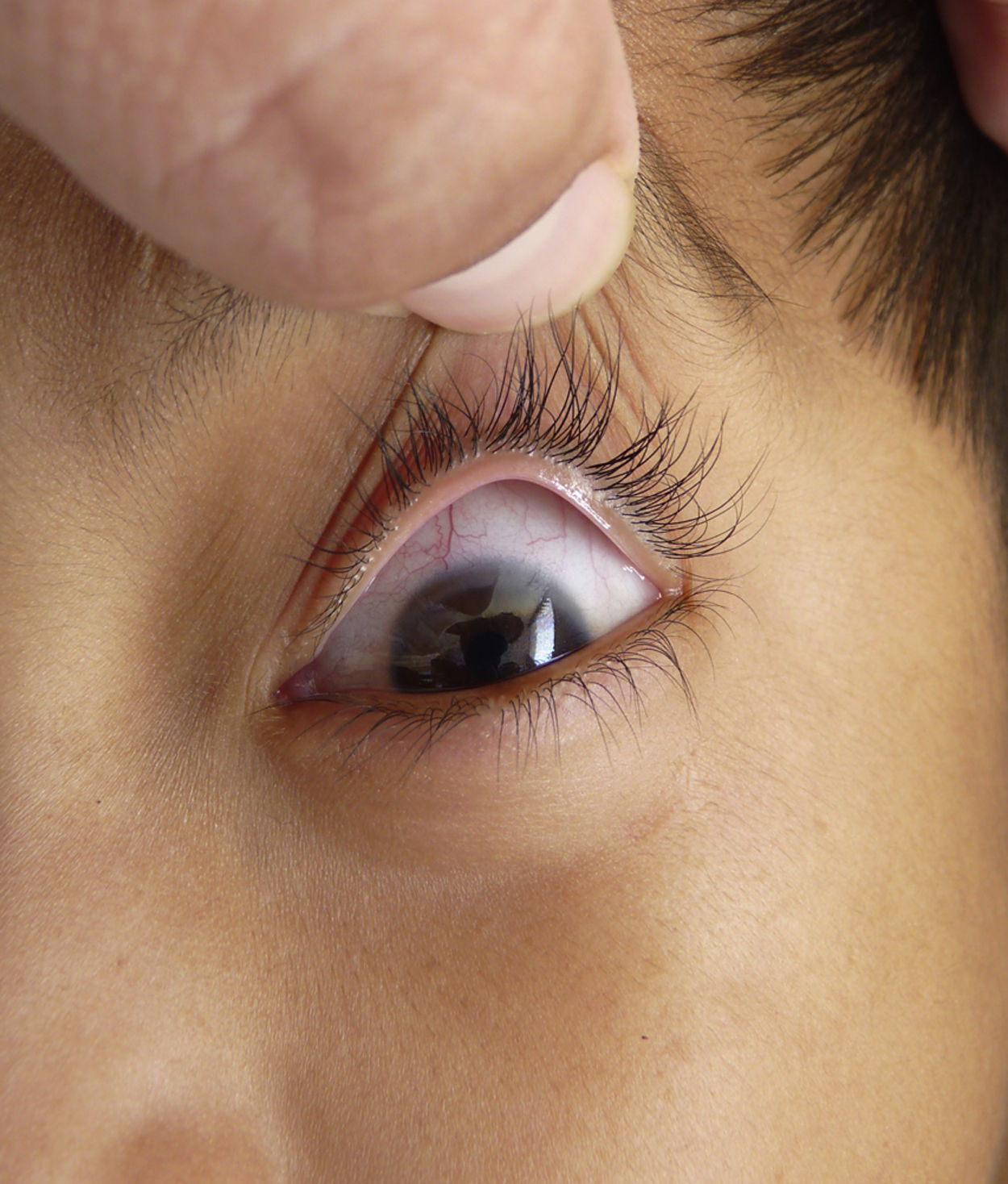
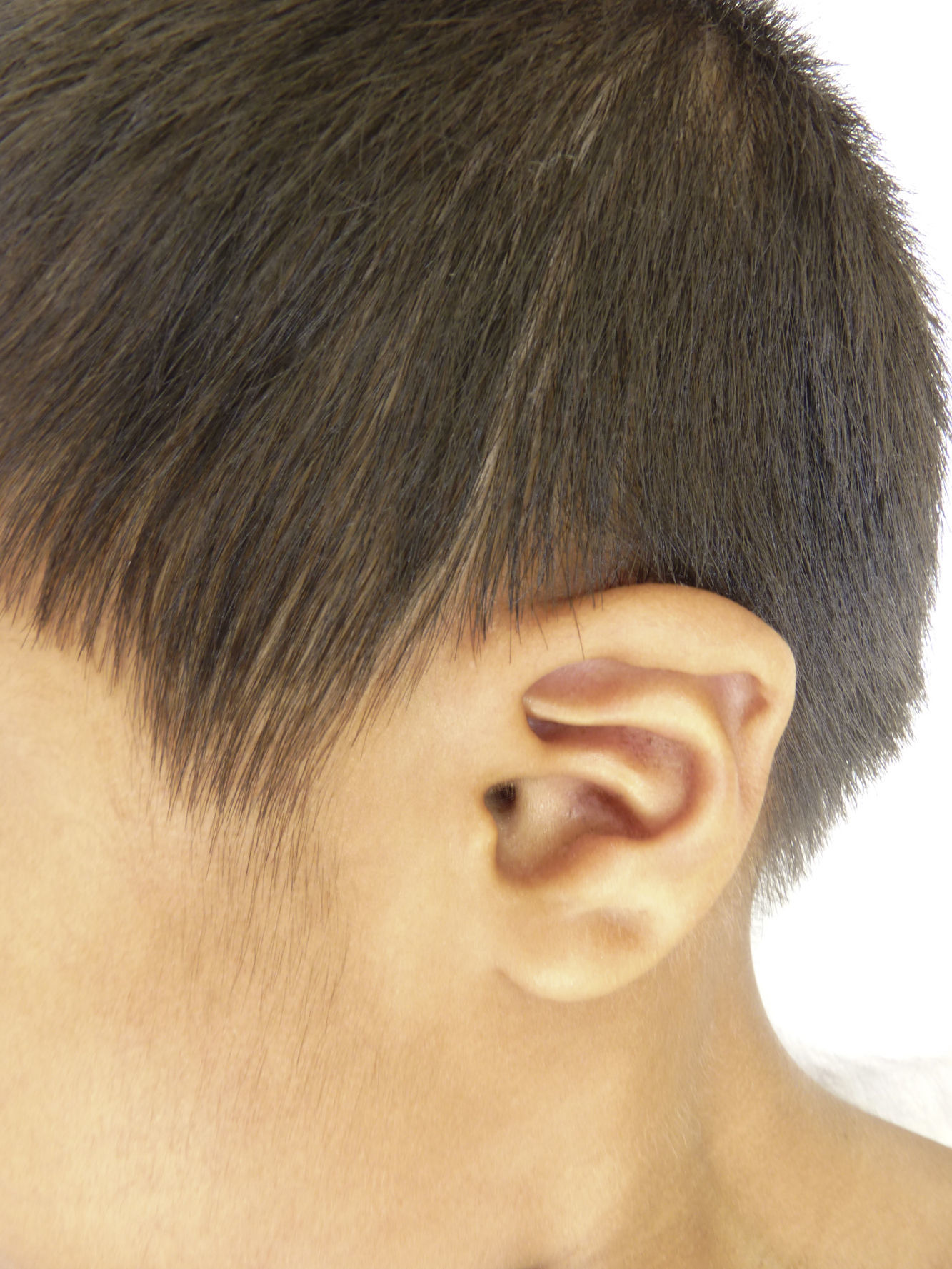
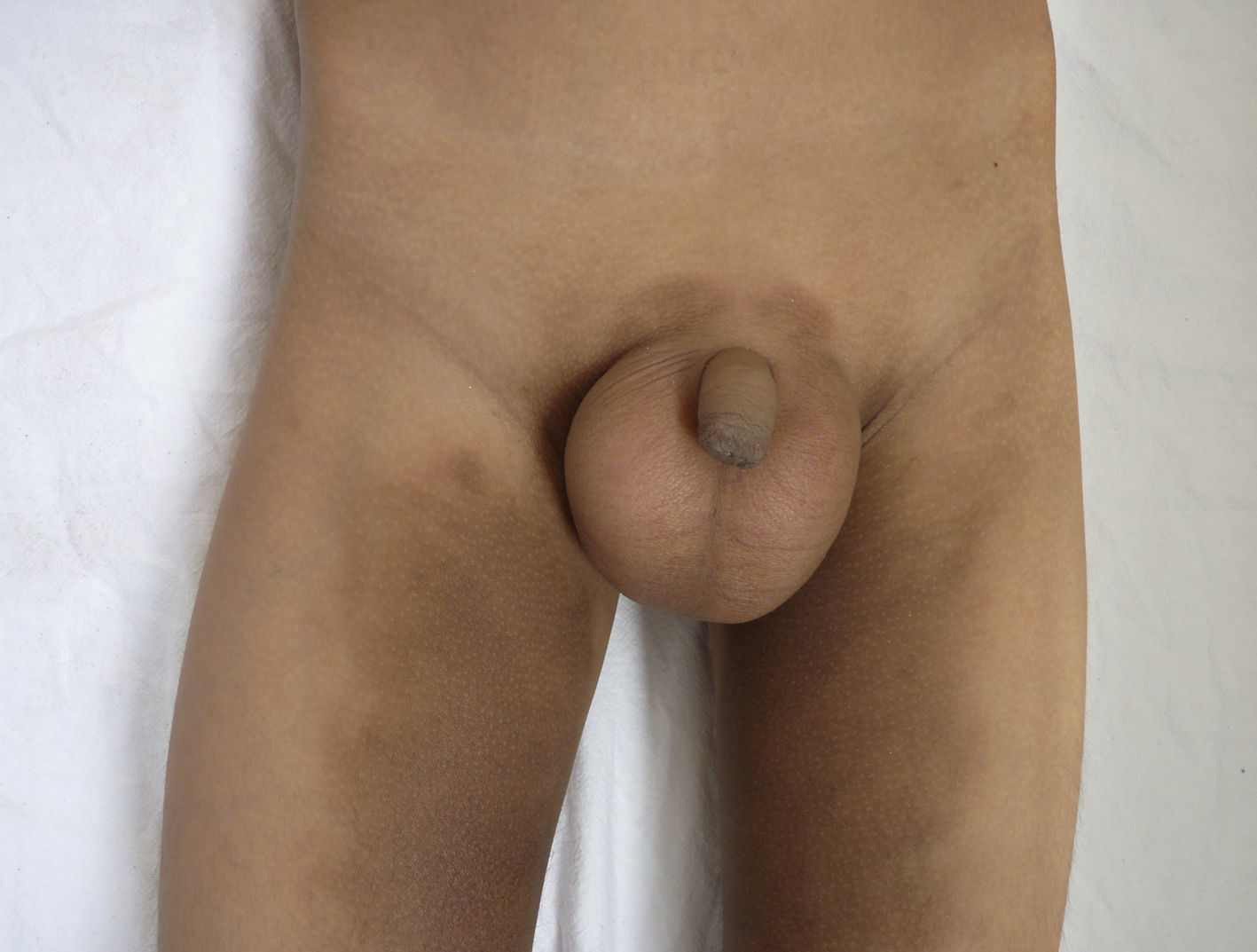
Otras características clínicas fueron palidez marcada, piel seca, hiperpigmentación e hipertricosis amplia en la región interna de ambos muslos y piernas (fig. 1). En la facies se observó ptosis palpebral bilateral, proptosis leve (fig. 2), halo senil bilateral (fig. 3), pabellones auriculares con hélix doblado (fig. 4), además de hiperplasia gingival y caries dental. Se detectaron adenopatías cervicales en cadena anterior, no dolorosas (no mayor de 1cm de diámetro). A la evaluación oftalmológica se halló dilatación de vasos esclerales y uveítis posterior. A la auscultación presentó ruidos cardiacos rítmicos, segundo ruido desdoblado, soplo sistólico grado ii/vi. Al examen de los genitales la longitud del pene fue de 3,2cm (<p10), el volumen de los testículos de 2cm3 según orquidómetro y vello pubiano Tanner 1. Se palparon 3 tumoraciones indoloras de consistencia firme, de 1,5×1cm en cada bolsa escrotal (fig. 5). Al examen de las extremidades fueron hallados cubitus valgus, leve deformidad en flexión de la región interfalángica proximal de ambas manos, tumefacción y limitación funcional en ambos tobillos, hallux valgus leve bilateral y pies planos.




A la evaluación antropométrica destacó peso de 19kg (–2,44 desviaciones estándar [DE]), talla de 110cm (–3,48DE), IMC de 15,7 (–0,09 DE), perímetro cefálico de 50cm (–1,28 DE), según tablas de la OMS.
En los exámenes de laboratorio se encontró anemia microcítica hipocrómica (hemoglobina: 7,8g/dl; volumen corpuscular medio: 57,7, hemoglobina corpuscular media: 16,0), hipergammaglobulinemia Ig A: 1.017UI (valor normal [VN]: 70-400), IgG: 3.789UI (VN: 700-1.600) e IgE: 76 UI (VN<52), velocidad de sedimentación globular aumentada (64 para VN<20mm/h), HDL bajo de 16mg/dl (VN>40mg/dl), perfil hormonal normal, glucosa e insulina en ayunas dentro de los límites normales para la edad, calcio y fósforo séricos dentro de los límites normales, cariotipo: 46 XY.
Los potenciales evocados auditivos detectaron ausencia de ondas en el lado izquierdo e hipoacusia neurosensorial derecha.
En las radiografías de la columna vertebral y los miembros se evidenció incremento de la radiolucidez compatible con osteopenia e hiperostosis en la radiografía de cráneo.
El ecocardiograma fue normal; en la ecografía testicular describen un aumento difuso del grosor y ecogenicidad de las partes blandas del escroto, con testículos de aspecto conservado. En la ecografía abdominal se observaron los ganglios mesentéricos de 7×3mm; hígado, bazo y riñones sin alteraciones.
El estudio anatomopatológico por biopsia escisional de tumoraciones de ambos testículos nos mostró tejido fibroadiposo con inflamación crónica granulomatosa severa, de predominio linfoplasmocitario, con abundantes macrófagos, CD45 y CD68 positivos en la inmunohistoquímica.
En la biopsia de piel se describió un aumento de grosor de la dermis, con depósitos de bandas de colágeno que se extienden desde la dermis hasta el tejido celular subcutáneo, infiltrado inflamatorio compuesto por linfocitos, células plasmáticas distribuidas de forma difusa en la lámina media, profunda y subcutis, engrosamiento septal en el tejido celular subcutáneo con moderado infiltrado linfoplasmocitario.
El secuenciamiento del gen SCL29A3 detectó la mutación homocigota c.1087 C>T (p.Arg363Trp; rs387907067), concluyente con el síndrome H. No se realizó el estudio molecular a los padres del niño.
En su evolución se evidenció incremento de lesiones dérmicas, así como de la deformidad de articulaciones interfalángicas de ambas manos y tobillos.
DiscusiónEl síndrome H fue descrito en 2008 como un trastorno autosómico recesivo en 10 pacientes, quienes procedían de familias palestinas consanguíneas1. El término fue elegido debido a las características de este trastorno con hiperpigmentación e hipertricosis, hepatoesplenomegalia, hipoacusia (característica de aparición precoz), anomalías del corazón (heart), hipogonadismo, talla baja (low height), hiperglucemia. Además se observó diabetes mellitus, contracturas en flexión y hallux valgus2,9–11.
El hallazgo clínico en la mayoría de los pacientes es hiperpigmentación cutánea e hipertricosis, que generalmente aparece en la parte medial de los muslos y en las crestas tibiales, con preservación de las rodillas, pero que puede ser más generalizada (tabla 1)1–3,5,7,9. Los primeros síntomas se pueden observar en el segundo año de vida, con linfadenopatía crónica, la cual puede ser generalizada o localizada (inguinal, cervical o axilar)3,12,13. La hipoacusia (manifestada como retraso del lenguaje) es una manifestación precoz, teniendo como promedio los 5,9 años de edad (nacimiento-14 años de edad)1,3,14. Otra de las características es el diagnóstico de diabetes mellitus, el cual puede aparecer tan precozmente como al año y medio de edad3,14,15. El promedio de edad de la aparición de hiperpigmentación es de 9,7 años de edad (1,5 meses a 27 años de edad)3.
Manifestaciones clínicas del síndrome H con mutación del gen SLC29A1
| Manifestaciones clínicas | N=79 | % | Caso reportado |
|---|---|---|---|
| Hiperpigmentación cutánea (con o sin hipertricosis e induración) | 54 | 68 | + |
| Contractura en flexión de dedos | 44 | 56 | + |
| Hipoacusia | 42 | 53 | + |
| Talla corta | 39 | 49 | + |
| Hepatomegalia | 34 | 43 | - |
| Esplenomegalia | 31 | 39 | - |
| Anomalías cardíacas (anormalidades pericárdicas) | 27 | 34 | - |
| Hallux valgus | 24 | 30 | + |
| Aumento de volumen y masas escrotales | 23 | 29 | + |
| Vasos esclerales dilatados, epiescleretis | 23 | 29 | + |
| Exoftalmos, proptosis, párpados con aumento de volumen | 22 | 28 | + |
| Venas varicosas | 22 | 28 | - |
| Ginecomastia (varones)a | 12 | 28 | - |
| Micropene (varones)a | 10 | 23 | + |
| Telangiectasia facial | 21 | 27 | - |
| Linfadenopatía | 19 | 24 | + |
| Diabestes mellitus insulinodependiente | 18 | 23 | - |
| Pie plano (deformidad de la piel) | 16 | 20 | + |
| Hipogonadismo | 13 | 16 | - |
| Azoospermia (varones)a | 9 | 9 | No evaluado |
| Malabsorción | 12 | 15 | - |
| Arco senil | 11 | 14 | + |
| Aumento de secreción de mucosa oral y nasal | 8 | 10 | - |
| Anormalidades en la médula ósea | 8 | 10 | No evaluado |
| Fracturas óseas y lesiones líticas | 7 | 9 | - |
| Artritis | 6 | 8 | + |
| Cambios ictiósicos | 6 | 8 | - |
| Anormalidades renales | 5 | 6 | - |
| Lipodistrofia glútea | 5 | 6 | - |
| Fiebre recurrente | 4 | 5 | + |
| Hidrocefalia/hipertensión intracraneal benigna/edema cerebral | 4 | 5 | - |
| Discapacidad intelectual leve | 4 | 5 | + |
| Hipertrigliceridemia | 3 | 4 | - |
| Hipertrofia gingival | 2 | 3 | + |
| Aumento de volumen de mejillas | 1 | 1 | + |
Estas lesiones son causadas por la infiltración de células plasmáticas de la dermis y región subcutánea. El transportador de nucleósidos hENT3 se expresa en los histiocitos, endotelio y linfocitos de la dermis normal humana. Mutaciones del gen SLC29A3 producen la proliferación anormal de histiocitos, alterando la respuesta inmune y aumentando la permeabilidad de los vasos sanguíneos y linfáticos. La infiltración y activación de linfocitos en la dermis produce inflamación y posterior hiperpigmentación. La presencia de histiocitos aberrantes en la vaina dérmica alrededor del folículo piloso puede llevar al crecimiento excesivo del vello en la piel en esta enfermedad16.
No hay clara correlación entre el fenotipo y el genotipo en el síndrome H. Los antecedentes genéticos, los genes modificadores y/o los factores ambientales pueden estar implicados en la determinación de la variabilidad fenotípica17.
El paciente que presentamos tiene los hallazgos clínicos compatibles con los casos publicados, formando parte del espectro de alteraciones del gen SLC29A3, excepto la hiperglucemia que no apareció hasta el momento. Es importante precisar que la mutación hallada en este paciente también se encontró en un niño de origen español de 13 años de edad, producto de un matrimonio no consanguíneo, teniendo como primeras manifestaciones la hiperpigmentación18.
Dentro del diagnóstico diferencial para el síndrome H tenemos enfermedades caracterizadas por cambios en la piel como esclerosis, hiperpigmentación e hipertricosis asociadas a manifestaciones sistémicas, tales como el síndrome Winchester y el síndrome de polineuropatía, organomegalia, endocrinopatía, proteína M y cambios en la piel (POEMS). En el síndrome Winchester se presenta con artropatía y facies tosca, sin hipoacusia.
El síndrome POEMS también se excluye en nuestro paciente debido a la ausencia de organomegalia, endocrinopatía y polineuropatía. Además, el patrón de los cambios cutáneos descritos no coincide con el síndrome Winchester o POEMS17.
Es importante aclarar que la mutación homocigota en el gen SCL29A3 también fue descrita como causa de disosteoesclerosis, la cual es una forma de osteopetrosis19.
ConclusionesEl síndrome H es una rara genodermatosis, de herencia recesiva autosómica, con manifestaciones sistémicas, causada por las mutaciones del gen SLC29A3 con amplio espectro fenotípico (clínicamente heterogenéneo). El hallazgo clínico principal, el cual orienta al diagnóstico es la hiperpigmentación acompañada de hipertricosis.
Los clínicos deben ser conscientes de las diversas manifestaciones de esta nueva condición descrita, para una identificación precoz, considerando su carácter crónico y evolutivo, así como la aparición por ejemplo de diabetes mellitus.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
