


El síndrome de trisomía 18 (T18) ocurre por la presencia de un cromosoma 18 extra completo en la mayoría de los casos. La prevalencia en recién nacidos oscila entre uno en 6.000 a uno en 8.000. Los afectados tienen una elevada mortalidad, solo el 4% supera el primer año de vida. Son pocos los casos reportados que superan los 5 años.
ObjetivoEl objetivo de este artículo es reportar un caso de T18 de larga sobrevida con características en la cavidad oral no descritas en la literatura, y aportar información a médicos y pediatras sobre la etiología, el fenotipo, la sobrevida y el consejo genético.
Reporte de casoPaciente de sexo femenino de 7 años con 2 cariotipos realizados en cultivo de linfocitos que mostraron 47,XX+18 en todas las metafases. Con talla y peso bajos, facies dismórficas, retardo severo del desarrollo psicomotor y cognitivo, imposibilidad para alimentarse, ausencia del lenguaje verbal, sordera neurosensorial, marcha atáxica, hipoplasia cerebelosa; genitales con labios mayores y menores hipoplásicos. En la cavidad oral paladar en forma de cúpula, macroglosia, no se observaron incisivos centrales superiores y primeros molares superiores e inferiores. En las radiografías se encuentran hallazgos de formación de las piezas dentales ausentes en la boca, concluyéndose erupción tardía.
ConclusionesEn los casos de T18 la mortalidad in útero y neonatal es alta, las características clínicas in útero y en recién nacidos han sido bien descritas. Dado que son pocos los casos que superan los 5 años el fenotipo aún está por establecerse. En la paciente aquí reportada se encontraron hallazgos en la cavidad oral no descritos en la literatura.
The trisomy 18 syndrome occurs due to the presence of an extra chromosome 18 in most cases. The prevalence in infants is estimated at 1:6000 to 1:8000. Those affected have a high mortality rate, only 4% may survive their first year of life. There are few reported cases exceeding five years of age.
ObjectiveThe aim of this paper is to report a case of trisomy 18 of long survival with oral cavity features not described in the literature, and to provide information to physicians and paediatricians about aetiology, phenotype, survival and genetic counselling.
Case reportA 7 year-old female patient with 2 karyotypes performed by lymphocyte culture showing 47XX+18 in all metaphases. She presented with growth deficiency, dysmorphic facies, severe psychomotor retardation and cognitive disability, inability to feed, lack of verbal language, sensorineural hearing loss, ataxia, cerebellar hypoplasia, and genitals with hypoplastic labia majora and minora. In the oral cavity: dome shaped palate, macroglossia, absence of upper central incisors and first upper and lower molars in mouth. X-ray findings showed formation of missing teeth, with late eruption being concluded.
ConclusionsIn cases of trisomy 18 syndrome there is an increased risk of neonatal and infant mortality. The clinical characteristics in utero and in neonates have been well described. Since few cases exceeding five years of age have been reported, the phenotype is yet to be established. In the case being reported we describe oral cavity findings not documented in the literature.
El síndrome de trisomía 18 (T18) o síndrome de Edwards es un trastorno cromosómico autosómico que se caracteriza por la presencia de un cromosoma 18 extra completo; además, se han reportado en la literatura casos de T18 en mosaico y de trisomía parcial. La prevalencia de esta trisomía en recién nacidos se calcula entre uno en 6.000 a uno en 8.000, siendo la segunda alteración cromosómica autosómica en frecuencia después de la trisomía 211. Esta prevalencia adquiere un valor más alto si se considera la muerte fetal y la interrupción voluntaria del embarazo después del diagnóstico prenatal, ascendiendo hasta uno en 2.500 o uno en 2.600. La frecuencia de la T18 aumenta con la edad materna2.
Los recién nacidos (RN) con T18 tienen una elevada mortalidad, aproximadamente el 50% viven más de una semana y alrededor del 5-10% viven más allá del primer año; pocos casos se han reportado que sobrevivan después de los 5 años3. Las características fenotípicas en mayores de 5 años más comunes son la talla y el peso bajos, la facies dismórfica, el retardo severo del desarrollo psicomotor y cognitivo, la imposibilidad para alimentarse, la ausencia del lenguaje verbal, los defectos del septo interventricular y la incapacidad para caminar por sí mismos2,4. Pocas anomalías han sido reportadas en la cavidad oral5.
Usualmente el diagnóstico del síndrome de T18 se sospecha en la etapa prenatal en función de la presencia de marcadores ecográficos y pruebas bioquímicas en el suero materno, o por anomalías anatómicas diagnosticadas en la ecografía prenatal, por lo cual se hace un procedimiento invasivo como biopsia de vellosidad corial, amniocentesis o cordocentesis para realizar un cariotipo o Fluorecense In Situ Hibridization (FISH) o hibridación genómica comparativa por microarreglos (mHGC)6–8; en la vida extrauterina se diagnostica usualmente con cariotipo bandas G en sangre periférica8.
El objetivo de esta publicación es reportar un caso de T18 de sobrevida inusual con características en la cavidad oral no descritas previamente en la literatura, y aportar información a médicos y pediatras en general sobre el fenotipo y la sobrevida de los afectados, elementos que podrían contribuir a la consejería genética.
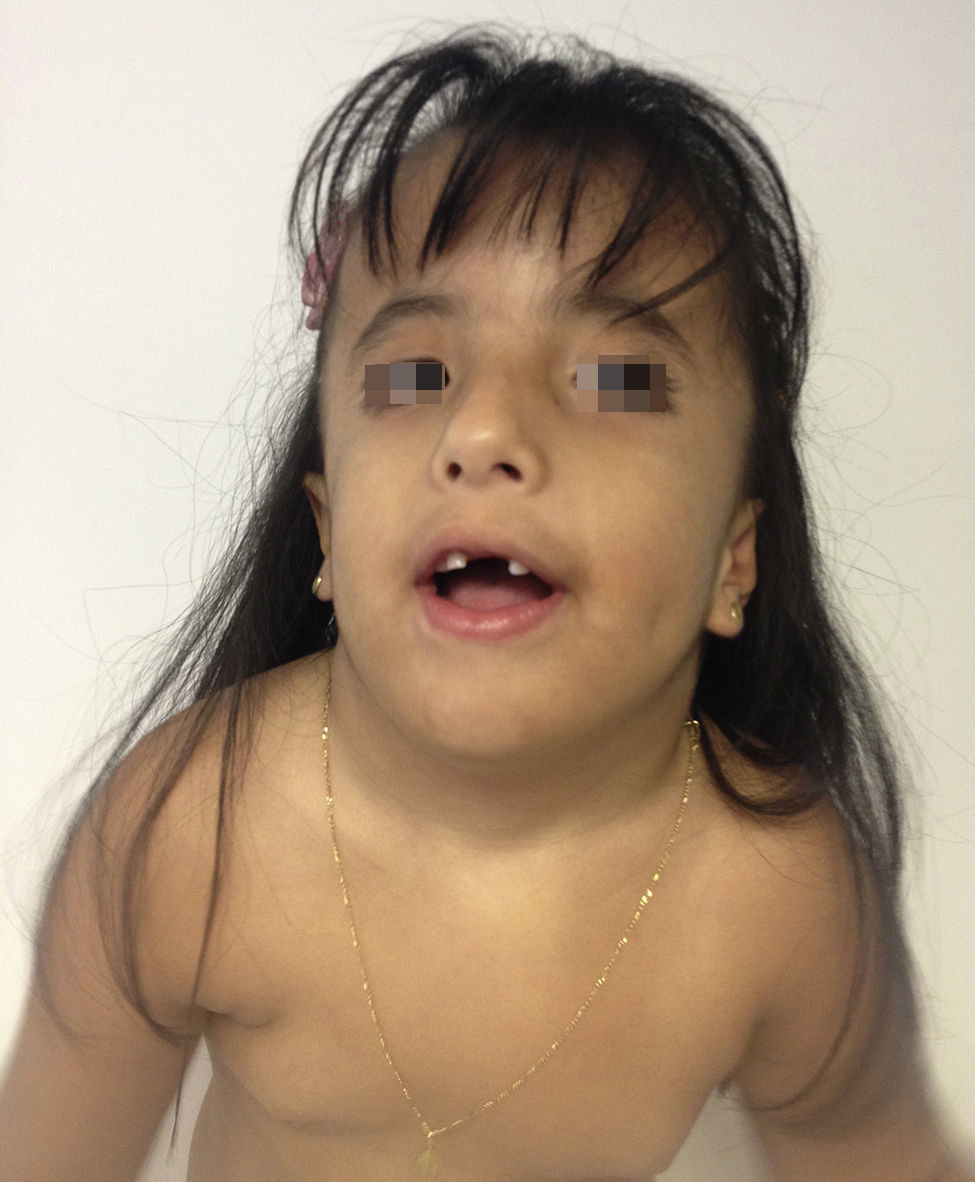
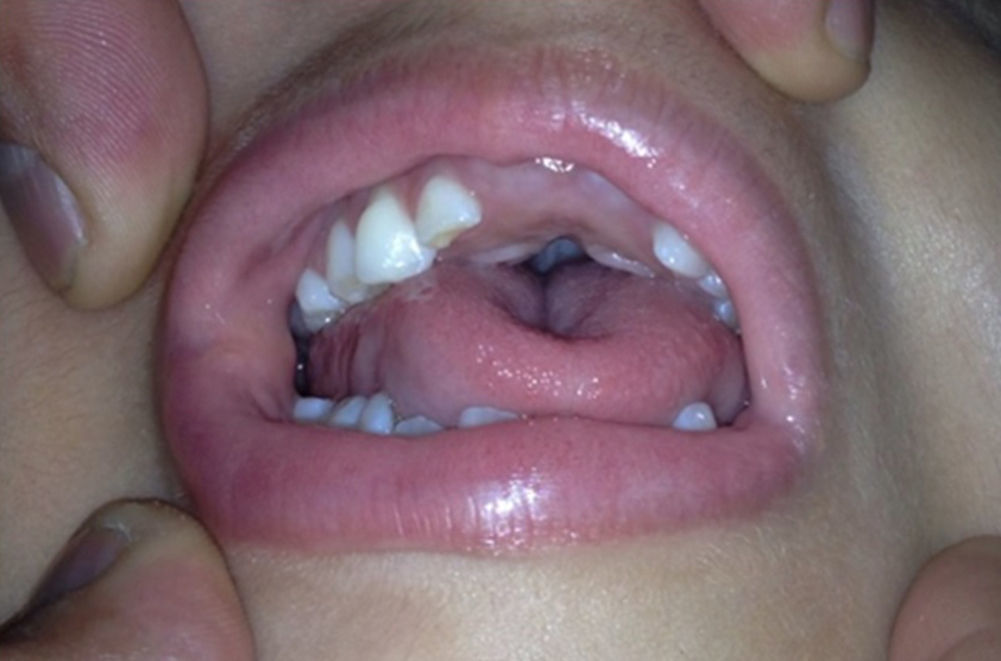
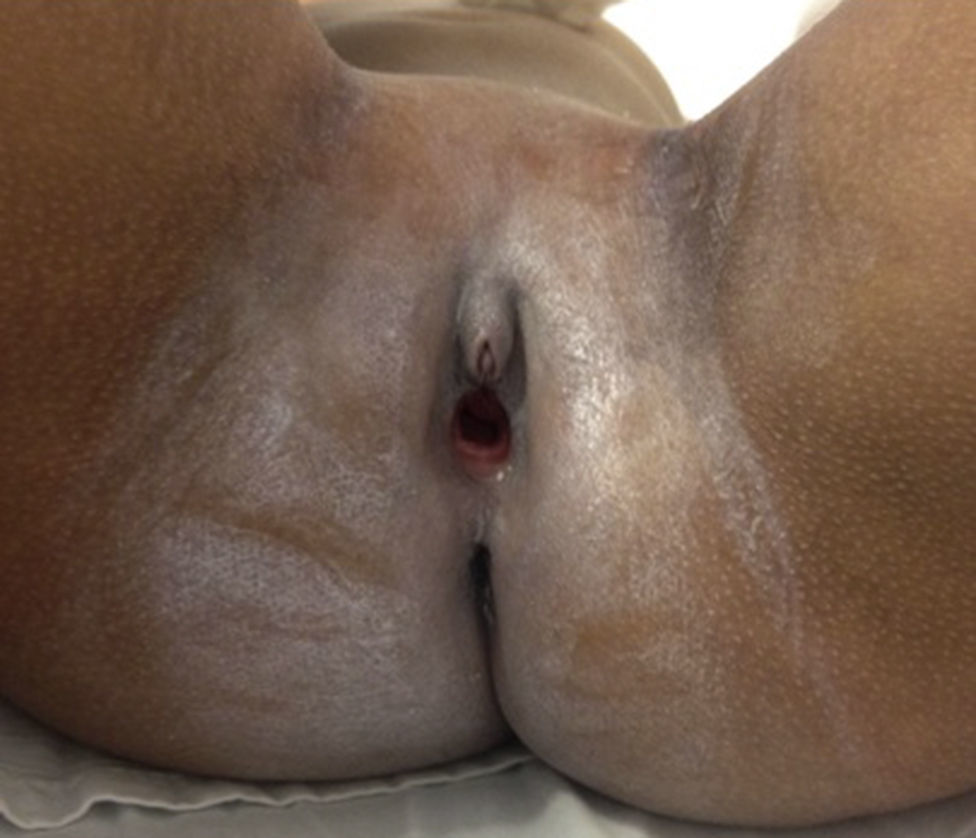
Caso clínicoPaciente de sexo femenino que consulta a los 7 años de edad en una clínica de dismorfología y genética. En esa evaluación la paciente presentó: talla 105cm (–2,48DS), peso 16,8 kg (-2 DS), perímetro cefálico 50cm (−1DS). Los hallazgos dismórficos observados al examen físico fueron: asimetría facial con microsomía facial derecha, depresión bitemporal, fisuras palpebrales inclinadas hacia abajo, estrabismo, alas de la nariz hipoplásicas, puente nasal alto, filtro nasal y surcos nasolabial y mentolabial poco marcados (fig. 1); en la cavidad oral se observó paladar profundo en forma de cúpula, macroglosia relativa con indentaciones en el borde de la lengua, atrición generalizada por bruxismo diurno; en la boca presentó 8 dientes superiores deciduos y 10 dientes inferiores, de estos últimos 4 correspondían a los incisivos permanentes, los demás eran deciduos; no se observaron incisivos centrales superiores y primeros molares superiores e inferiores (fig. 2). Además, la paciente presentó pectus carinatum insinuado, hipoplasia de radio y ulna, hipoplasia del tenar, pliegue palmar único, braquidactilia del quinto y primer dedo, hipoplasia de la uña del quinto dedo, halux ancho bilateral; labios menores y mayores hipoplásicos (fig. 3).
.")
Cara de la paciente a los 7 años. Se puede observar: asimetría facial con microsomía facial derecha, depresión bitemporal, fisuras palpebrales inclinadas hacia abajo, estrabismo, alas de nariz hipoplásicas, puente nasal alto, filtro nasal y surcos nasolabial y mentolabial poco marcados, ausencia de incisivos centrales (fotografía con autorización).
.")
Cavidad oral de la paciente a los 7 años. Se observa la forma de cúpula del paladar, macroglosia relativa con indentaciones en el borde de la lengua, facetas de desgaste oclusal principalmente en los dientes posteriores. No se observaron incisivos centrales superiores (fotografía con autorización).
.")
Genitales de la paciente a los 7 años. Se observa ausencia de labios menores, labios mayores hipoplásicos, himen anular amplio no festoneado; clítoris de tamaño normal, pero que aparenta clitoromegalia por desproporción con los labios mayores y menores (fotografía con autorización).
La paciente es hija de padres no consanguíneos; en las familias no había antecedentes de enfermedades médicas agudas, crónicas, infecciosas o genéticas relevantes. La madre se embarazó de la paciente a los 28 años, siendo su primera gestación; hizo control prenatal, no encontraron enfermedades o exposiciones a teratógenos y le realizaron 3 ecografías, a las 9 y 23 semanas de gestación, que no mostraron alteraciones; en la ecografía de las 32 semanas se observó megacisterna magna de 16,9mm y restricción de crecimiento intrauterino. Le realizaron cesárea por sufrimiento fetal agudo. Al nacimiento encontraron varios hallazgos dismórficos y le realizaron el cariotipo con bandas G realizado en el cultivo de linfocitos, con un nivel de resolución de 660 bandas, que mostró 47, XX+18 en el 100% de las 30 metafases observadas.
A lo largo de la vida la paciente presentó retraso global del desarrollo. A los 7 años no controla esfínteres, no pronuncia silabas, solo emite ruidos, camina con ayuda, con marcha inestable, atáxica. Además, se le diagnosticó sordera neurosensorial bilateral con potenciales auditivos.
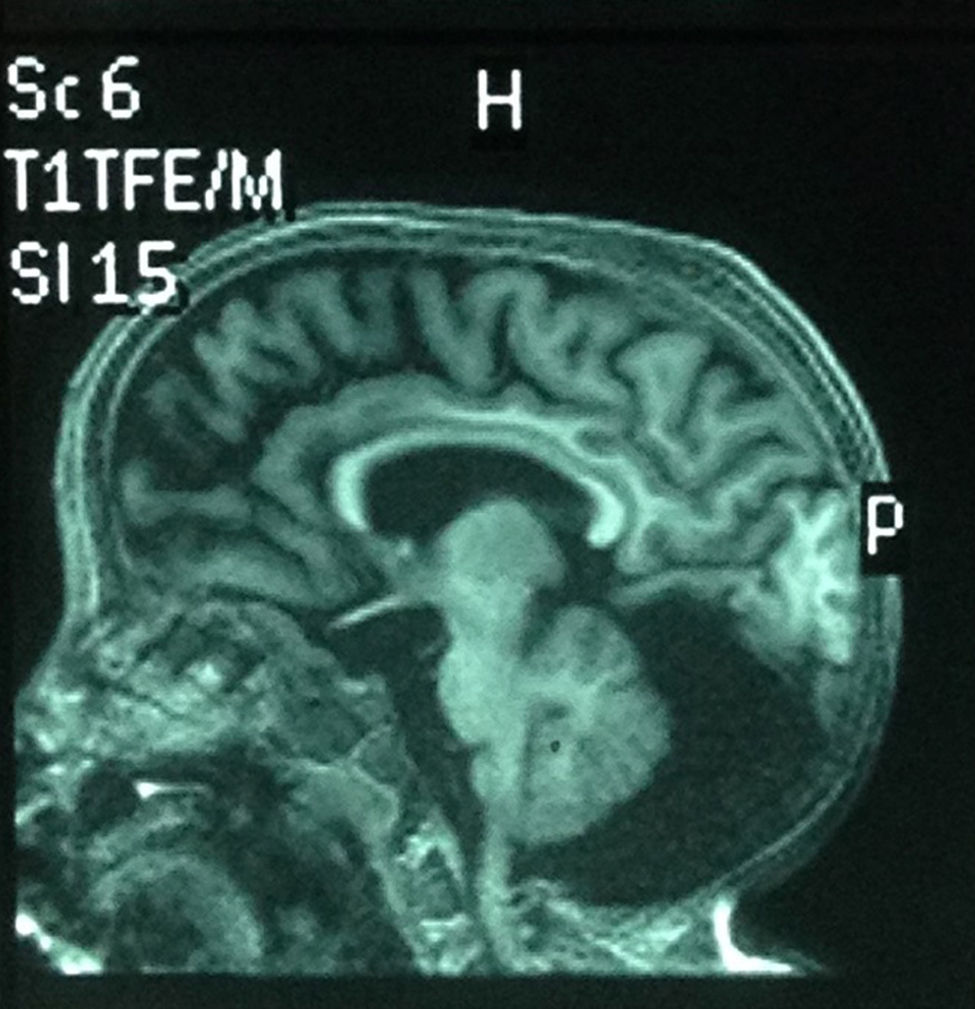
Comenzó con convulsiones a los 4 años, manejada con ácido valproico, y tiene en el último año un único episodio convulsivo. EEG: anormal por la presencia de brotes paroxísticos de ondas lentas derechas, con ocasional espejo y énfasis occipital sobre disfunción cortical de base. La TAC cerebral a los 4 años mostró megacisterna magna y disminución en el diámetro cerebral. En la resonancia magnética nuclear (RMN) realizada a los 6 años se observaron cambios atróficos difusos, quiste aracnoideo retro y supracerebeloso; además, hipoplasia del vérmix cerebeloso sin comunicación con el iv ventrículo, cerebelo atrófico (fig. 4). Además, presenta reflujo gastroesofágico severo que se diagnosticó con esófago-gastro-dueodenograma; tiene gastrostomía.

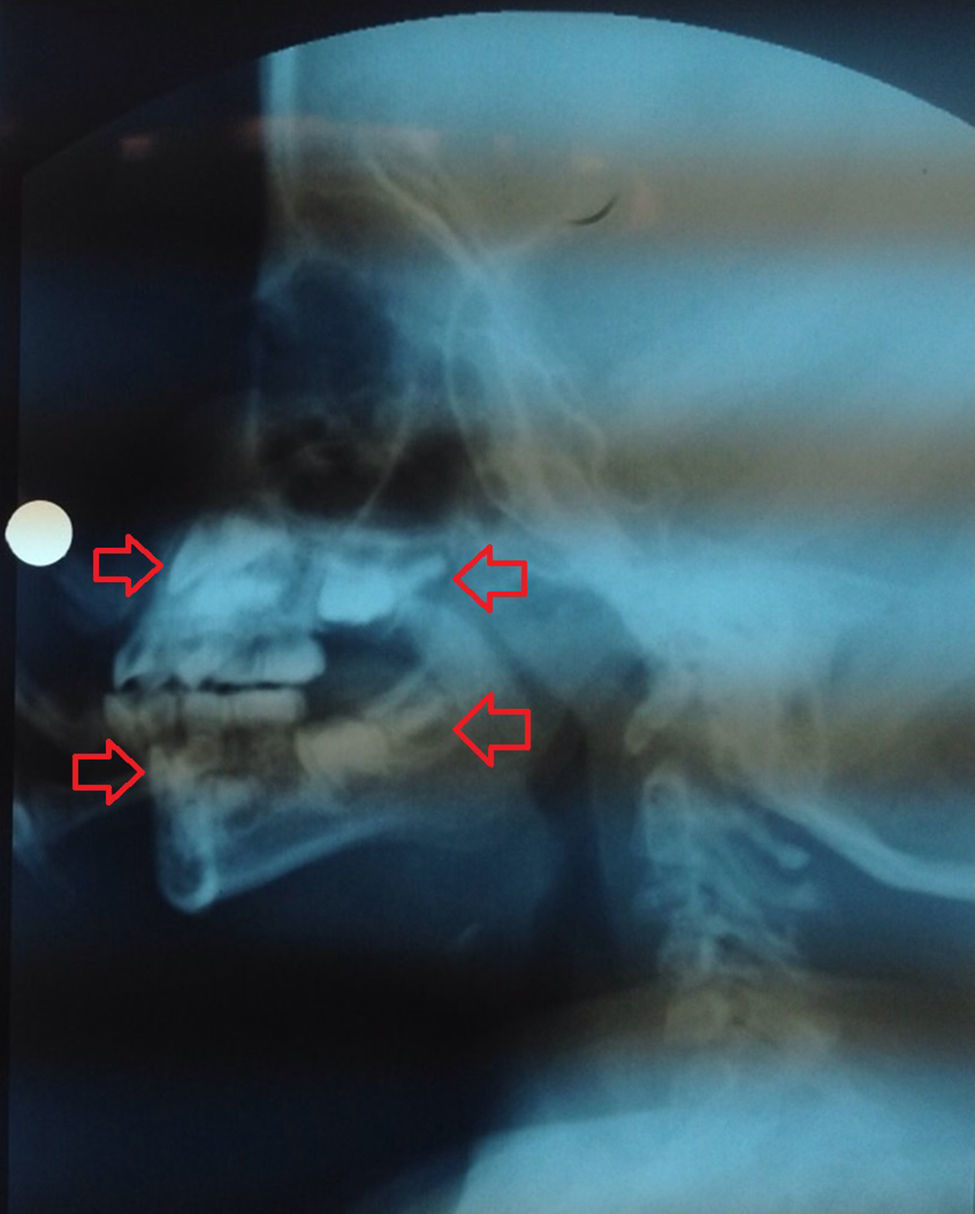
En radiografía lateral de cráneo, tomada dentro del estudio de deglución, se observó evidencia de la formación de la corona de los primeros molares superiores e inferiores y de los incisivos superiores; considerando la edad de la paciente estos dientes deberían estar en proceso de erupción, es decir, en un estadio de desarrollo más avanzado (fig. 5); En la ortopantomografía (de mala calidad por pobre colaboración de la paciente) se observaron los 2/3 coronales de las raíces de los primeros molares y la formación de la corona de los segundos molares de ambos lados.

Radiografía lateral de cráneo. Se destaca en flechas rojas la formación de la corona de los incisivos superiores e inferiores y de los primeros molares superiores e inferiores. Debido a la sobreproyección de estructuras no es posible establecer con certeza la lateralidad de los dientes observados en la radiografía.
En el ecocardiograma en periodo neonatal y en los controles anuales se observó una comunicación interauricular sin repercusión hemodinámica; en la última evaluación a los 6 años y 10 meses no se encontró. La ecografía abdominopélvica mostró riñones, vías urinarias, útero y ovarios sin alteración.
A los 6 años repiten el cariotipo, dado que son muy pocos los casos en la literatura que han superado el año de vida con esa presentación cromosómica; el nuevo cariotipo con bandas G con resolución de 660 bandas mostró 47, XX+18 en 30 metafases, realizado en cultivo de linfocitos.
La madre de la paciente aceptó y firmó consentimiento informado para toma de fotografías y tomar datos de la historia clínica para la publicación del caso.
DiscusiónLa T18 ocurre por la presencia de un cromosoma 18 adicional al par 18 usual en todas las células; también se puede presentar en mosaicos cuando la T18 no está presente en todas la células del mismo individuo, o por trisomía parcial, en la que existe un fragmento extra del cromosoma 18, que puede ser un brazo completo (usualmente el largo 18q) o una región de este1. La T18 completa en todas las células es la forma más frecuente, representa aproximadamente el 94% de los casos2. El diagnóstico se confirma con la realización de cariotipo, y el más usado es con la tinción de Giemsa, aunque también se puede realizar FISH o hibridación genómica comparativa por microarreglos, todos los cuales se pueden realizar en diagnóstico prenatal o posnatal8. La probabilidad de tener un hijo con T18 aumenta con la edad materna2.
La prevalencia de la T18 varía entre los grupos poblacionales a nivel mundial. Globalmente se estima en 1:6000 nacidos vivos, observándose que los afectados de mayor proporción son los de sexo femenino; sin embargo, se debe considerar que las interrupciones voluntarias del embarazo se han realizado en mayor medida con fetos masculinos. Por otra parte, la supervivencia es baja y solo uno de cada 10 a 20 neonatos alcanza el primer año de vida, siendo las mujeres las que han mostrado mayor tiempo de supervivencia2. Las principales causas de muerte son las cardiopatías, la insuficiencia cardiaca, la apnea central, la insuficiencia respiratoria y la broncoaspiración, entre otros factores3,9,10. La paciente aquí reportada no ha presentado ninguna de estas enfermedades, lo que explica su sobrevida.
In útero el síndrome de T18 se sospecha en función de la identificación de hallazgos ecográficos. El aumento en la translucencia nucal y la ausencia de hueso nasal (también usados en el síndrome de Down y el síndrome de Patau) se observan en el 66% de los fetos con T1811,12. Otros marcadores ecográficos, como flujo reverso del ductus venoso y regurgitación en la válvula tricuspídea, aumentan la detección hasta un 83,3%12. Algunos protocolos integran marcadores ecográficos y bioquímicos en el primer trimestre y pruebas bioquímicas en el segundo trimestre, alcanzando tasas de detección hasta del 78%13,14; sin embargo, esta prueba de tamizaje integrada está en desuso.
Los fetos con T18 pueden presentar anomalías anatómicas únicas o múltiples. Algunas se pueden detectar en la ecografía del primer trimestre, pero con mayor probabilidad de observarse en el segundo o tercer trimestre del embarazo; estas son: onfalocele, postura anormal de las manos (desviación radial o ulnar), mega vejiga, anormalidades cardiacas (usualmente las detectables en la vista de 4 cámaras), megacisterna magna, retardo en el crecimiento intrauterino (siendo frecuente su inicio en el segundo trimestre del embarazo), polihidramnios, cráneo en forma de fresa, quiste de plexo coroideo, dedos superpuestos (conocido como mano trisómica, desviación de los dedos hacia la línea media), arteria umbilical única7,15,16. A la paciente aquí reportada no se le encontraron anomalías en la ecografía de las 9 y 23 semanas, pero le fue detectada una megacisterna magna y un retardo del crecimiento intrauterino a las 32 semanas. Estos hallazgos han sido reportados en los casos de T18.
Dado que el pronóstico de los recién nacidos (RN) con T18 es pobre, los estudios de sobrevida son consistentes. En promedio el 42% llegan vivos a la primera semana; el 29% al primer mes, el 12% a los 3 meses y el 8% a los 6 meses. Al año sobrevive alrededor del 4%17; además, como el pronóstico neurológico a largo plazo es pobre, a los RN con T18 suelen no realizárseles grandes procedimientos médicos o quirúrgicos en el periodo neonatal, los RN siguen la historia natural de la enfermedad con una mortalidad muy alta. Sin embargo, se ha descrito que cuando al RN se le realizan los protocolos usuales para cada una de las anomalías congénitas (como a otros RN sin T18) y es manejado en la unidad de cuidados intensivos la sobrevida mejora. La media de supervivencia alcanza los 152 días, el 88% alcanza la primera semana, el 83% el primer mes y el 25% llega vivo al año. Las maniobras médicas suelen incluir medidas de resucitación, ventilación mecánica, productos sanguíneos, nutrición parenteral, cirugías paliativas o correctivas gastrointestinales, alcanzar la nutrición enteral y manejo médico de los defectos cardiacos, todas las intervenciones con el consentimiento de los padres, a los que se les debe dar la información desde que el diagnóstico se hace in útero9,17.
Otros casos anecdóticos han sido reportados, en donde se han realizado cirugías correctivas de anomalías congénitas mayores como hernias diafragmáticas, onfalocele y defectos del tubo neural, y la sobrevida mejoró17.
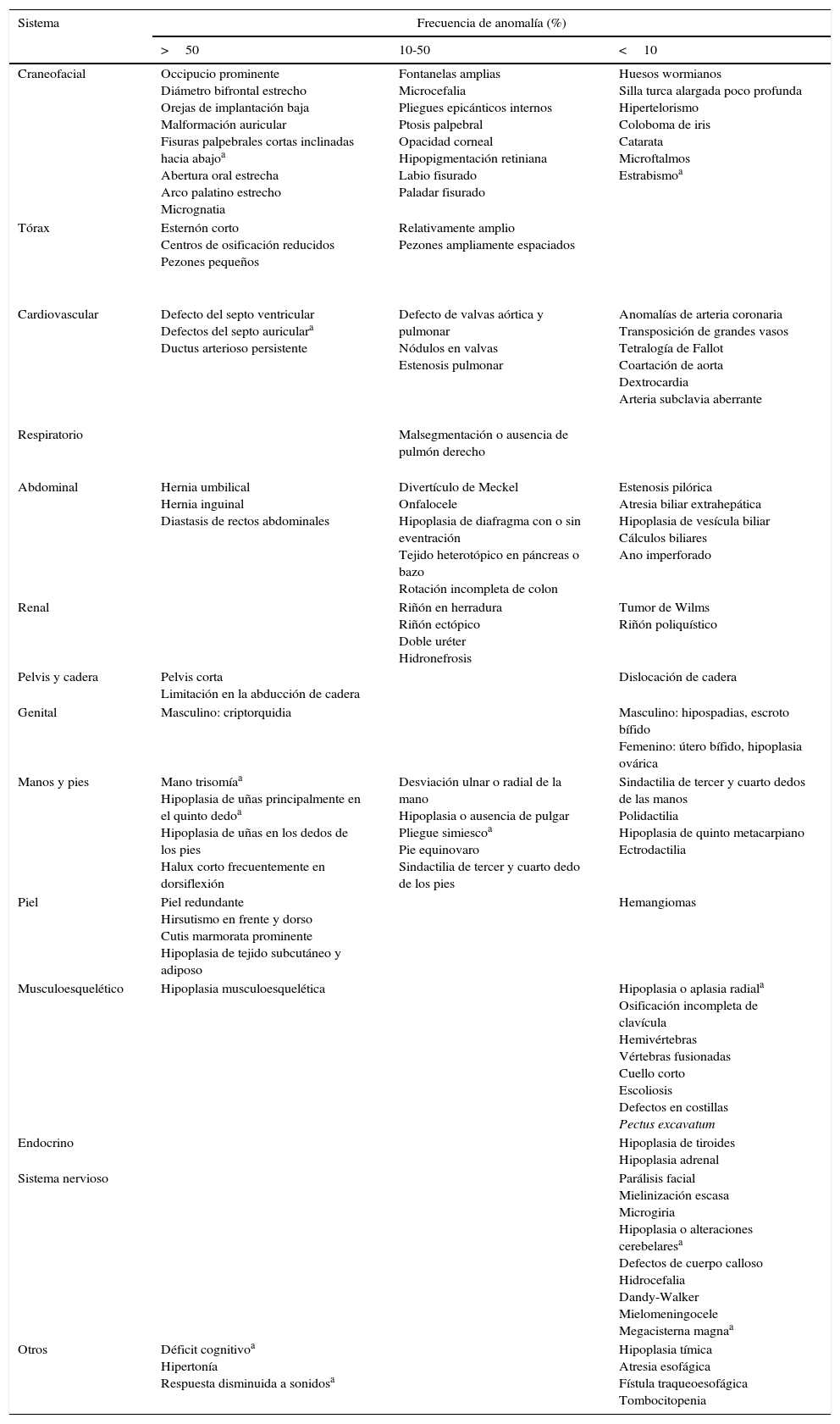
Los hallazgos fenotípicos más frecuentemente observados en pacientes con T18 RN o infantes se pueden observar en la tabla 1. Son pocos los reportes de sobrevida después de los 5 años5,18–20; en estos las características encontradas consistentemente fueron: talla y peso bajos, facies dismórfica, retardo severo del desarrollo psicomotor y cognitivo, imposibilidad para alimentarse, ausencia del lenguaje verbal, defectos del septo interventricular e incapacidad para caminar por sí mismos21; como hallazgos especiales se han reportado en estos pacientes cardiomiopatÍa hipertrófica20 y tumor de Wilms19. Además, en nuestra paciente se encontró sordera neurosensorial y los genitales con labios hipoplásicos. Uno de los hallazgos más representativos de la paciente fue la hipoplasia cerebelosa, que explica la marcha atáxica que le genera incapacidad para caminar sin ayuda. No presentó grandes defectos cardiovasculares o cerebrales, por lo cual se puede explicar la sobrevida, además de por el apoyo médico y familiar asistido a la paciente.
Frecuencia de anomalías encontradas en afectados con T18 y observadas en la paciente aquí reportada
| Sistema | Frecuencia de anomalía (%) | ||
|---|---|---|---|
| >50 | 10-50 | <10 | |
| Craneofacial | Occipucio prominente Diámetro bifrontal estrecho Orejas de implantación baja Malformación auricular Fisuras palpebrales cortas inclinadas hacia abajoa Abertura oral estrecha Arco palatino estrecho Micrognatia | Fontanelas amplias Microcefalia Pliegues epicánticos internos Ptosis palpebral Opacidad corneal Hipopigmentación retiniana Labio fisurado Paladar fisurado | Huesos wormianos Silla turca alargada poco profunda Hipertelorismo Coloboma de iris Catarata Microftalmos Estrabismoa |
| Tórax | Esternón corto Centros de osificación reducidos Pezones pequeños | Relativamente amplio Pezones ampliamente espaciados | |
| Cardiovascular | Defecto del septo ventricular Defectos del septo auriculara Ductus arterioso persistente | Defecto de valvas aórtica y pulmonar Nódulos en valvas Estenosis pulmonar | Anomalías de arteria coronaria Transposición de grandes vasos Tetralogía de Fallot Coartación de aorta Dextrocardia Arteria subclavia aberrante |
| Respiratorio | Malsegmentación o ausencia de pulmón derecho | ||
| Abdominal | Hernia umbilical Hernia inguinal Diastasis de rectos abdominales | Divertículo de Meckel Onfalocele Hipoplasia de diafragma con o sin eventración Tejido heterotópico en páncreas o bazo Rotación incompleta de colon | Estenosis pilórica Atresia biliar extrahepática Hipoplasia de vesícula biliar Cálculos biliares Ano imperforado |
| Renal | Riñón en herradura Riñón ectópico Doble uréter Hidronefrosis | Tumor de Wilms Riñón poliquístico | |
| Pelvis y cadera | Pelvis corta Limitación en la abducción de cadera | Dislocación de cadera | |
| Genital | Masculino: criptorquidia | Masculino: hipospadias, escroto bífido Femenino: útero bífido, hipoplasia ovárica | |
| Manos y pies | Mano trisomíaa Hipoplasia de uñas principalmente en el quinto dedoa Hipoplasia de uñas en los dedos de los pies Halux corto frecuentemente en dorsiflexión | Desviación ulnar o radial de la mano Hipoplasia o ausencia de pulgar Pliegue simiescoa Pie equinovaro Sindactilia de tercer y cuarto dedo de los pies | Sindactilia de tercer y cuarto dedos de las manos Polidactilia Hipoplasia de quinto metacarpiano Ectrodactilia |
| Piel | Piel redundante Hirsutismo en frente y dorso Cutis marmorata prominente Hipoplasia de tejido subcutáneo y adiposo | Hemangiomas | |
| Musculoesquelético | Hipoplasia musculoesquelética | Hipoplasia o aplasia radiala Osificación incompleta de clavícula Hemivértebras Vértebras fusionadas Cuello corto Escoliosis Defectos en costillas Pectus excavatum | |
| Endocrino | Hipoplasia de tiroides Hipoplasia adrenal | ||
| Sistema nervioso | Parálisis facial Mielinización escasa Microgiria Hipoplasia o alteraciones cerebelaresa Defectos de cuerpo calloso Hidrocefalia Dandy-Walker Mielomeningocele Megacisterna magnaa | ||
| Otros | Déficit cognitivoa Hipertonía Respuesta disminuida a sonidosa | Hipoplasia tímica Atresia esofágica Fístula traqueoesofágica Tombocitopenia | |
Pacientes con T18 en mosaico han sido descritos con un fenotipo variable, algunos con una mejor sobrevida que la descrita para pacientes con trisomía completa3,22. A la paciente aquí reportada se le realizaron 2 cariotipos, uno al nacimiento y otro a los 6 años de edad, los cuales mostraron la T18 en todas las metafases observadas; sin embargo, no es posible descartar que esta paciente tenga una T18 en mosaico debido a que la observación realizada en los cariotipos no llegó a 100 metafases; además, no se le realizó cariotipo en tejidos diferentes a cultivo de linfocitos, como piel o mucosas. Este diagnóstico también puede descartarse al realizar FISH para cromosoma 18 en biopsia de mucosa bucal, piel o sangre, analizando más de 200 células. Estos estudios complementarios no se le realizaron a la paciente dado que no se dispone de ellos a través de su servicio de salud.
En la literatura revisada se encontraron pocas descripciones del sistema estomatognático en RN, infantes o mayores de 5 años20. En la paciente aquí reportada se observaron hallazgos característicos de los respiradores orales. Las facetas de desgaste que se observan en la dentición temporal de la paciente sugieren una constante actividad de los músculos masticatorios, especialmente de maseteros y pterigoideo interno, sugiriendo esto que el problema de la deglución en los pacientes con T18 no está relacionado con déficit en los músculos de la masticación. En contraste con lo descrito por Ribeiro et al.5, la forma del paladar de la paciente es de cúpula y no presenta fisura palatina, sin embargo, esta característica puede asociarse con alteraciones en el proceso de fusión de los procesos palatinos durante la formación del paladar secundario.
Considerando la edad de la paciente, en boca deberían estar presentes los primeros molares permanentes y los incisivos centrales superiores. En las radiografías tomadas a la paciente se evidencian hallazgos relacionados con el desarrollo dental de los dientes no observados en boca, sugiriendo erupción tardía23. En la literatura revisada se encuentran descritas características de la cavidad oral de pocos pacientes5. Pese a que se mencionan múltiples anomalías en la dentición, no se menciona retraso o alteración en la secuencia de erupción, por lo que proponemos que la erupción tardía es un hallazgo no descrito previamente en la T18, y que debe ser evaluado en todos los pacientes con esta condición por un odontopediatra.
En los casos de diagnóstico prenatal de T18, en RN o en infantes, se debe brindar asesoría a la familia sobre las probabilidades de muerte in útero o en RN a edades específicas, y sobre el pronóstico neurológico, ya que deben tomar decisiones sobre la finalización voluntaria del embarazo (según la legislación de cada país), reanimación y realización de cirugías invasivas; cuando el paciente sobrevive después del primer mes es necesario que sea manejado por un grupo interdisciplinario, interviniendo de manera consensuada con los padres, para que trate las enfermedades específicas que presente cada niño con el fin de lograr la mejor calidad de vida posible, haciendo énfasis en la alimentación, probablemente por gastrostomía2.
También se debe proporcionar consejo genética, en el que se explique que para una pareja con un niño con T18 libre y completa la probabilidad de repetición en el siguiente embarazo es del 1%2,3. Los casos en que la T18 es parcial es necesario realizar cariotipo a los padres para descartar que sean portadores de traslocación balanceada que incluya el segmento trisómico, pues en estos casos la probabilidad de repetición es mayor2.
ConclusiónSe reporta el caso de una paciente con síndrome de T18 en el 100% de las metafases observadas en cultivo de linfocitos con inusual sobrevida —7 años—, lo que hace plantear la posibilidad de mosaico. Son muy pocos los casos reportados que sobrepasan los 5 años. En los casos de T18 la mortalidad in útero y neonatal es alta, no obstante, esta mejora cuando las anomalías congénitas son menores, se da soporte cuando se requiere en la unidad de cuidados intensivos y se realizan cirugías paliativas o correctivas. Las características clínicas in útero y en RN han sido bien descritas. En pacientes mayores de 5 años el fenotipo aún está por delimitarse, no obstante un rasgo constante en los pocos pacientes descritos es retraso mental de moderado a severo. Este artículo aporta información sobre hallazgos en la cavidad oral no descritos previamente en la literatura.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
