




La deleción de la región cromosómica 1p36 es una de las anomalías subteloméricas más frecuentes y causa rasgos dismórficos distintivos. Por otro lado, la trisomía distal del brazo corto del cromosoma 6 es una anormalidad cromosómica poco frecuente de fenotipo variable.
ObjetivoPresentar el caso de un paciente con ambas alteraciones cromosómicas, y resaltar la vigencia e importancia del cariotipo como herramienta diagnóstica en dismorfología.
Caso clínicoLactante de 2 meses de edad con múltiples anomalías craneofaciales, hemangioma en la nuca, fosita sacra, acortamiento rizomélico, pies y manos pequeños, criptorquidia unilateral izquierda e hipotonía. Además, antecedente de restricción del crecimiento intrauterino. Producto del octavo embarazo de una mujer G8A7C1 de 28 años. Con estos hallazgos inespecíficos en el fenotipo se solicitó cariotipo que mostró una deleción parcial de 1p36.1 y una trisomía parcial de cromosoma 6p.
ConclusiónEl cariotipo convencional sigue siendo una herramienta importante para el etiológico en pacientes con anomalías congénitas (múltiples), mostrando en este caso una deleción parcial de 1p36.1 y una trisomía parcial de cromosoma 6p, alteraciones cromosómicas estructurales.
The deletion of chromosomal region 1p36 is one of the most common sub-telomeric microdeletion syndromes and has distinctive dysmorphic features. On the other hand, partial trisomy of the short arm of chromosome 6 is a rare chromosomal abnormality with a variable phenotype.
ObjectiveTo report a case with both chromosome abnormalities, and to highlight the importance of the karyotype as a diagnostic tool in dysmorphology.
Clinical caseThe case of is presented of a two month-old infant with several craniofacial anomalies, neck haemangioma, sacral pit, rhizomelic shortening, small hands and feet, left unilateral cryptorchidism, and hypotonia. The infant also suffered intrauterine growth restriction and is the product of the eighth pregnancy of a 28 years old woman. Due to the unspecific findings in phenotype, a karyotype was requested, which showed a partial deletion of 1p36.1 and a partial trisomy of chromosome 6.
ConclusionThe development of new techniques in molecular biology has improved diagnostic possibilities in medical genetics. However, the traditional karyotype remains as an important diagnostic tool in patients with multiple congenital anomalies.
La monosomía 1p36 es el síndrome de microdeleción terminal más frecuente, con una prevalencia de uno por cada 5.000 recién nacidos1. Su fenotipo comprende la discapacidad intelectual, retraso del desarrollo, hipotonía, epilepsia, obesidad, hiperfagia y trastornos de la conducta2. Otras características comúnmente descritas incluyen anomalías faciales, hipoacusia sensorineural, alteraciones visuales, cardiopatías congénitas, cardiomiopatía, problemas gastrointestinales y pubertad precoz3,4.
La trisomía distal del brazo corto del cromosoma 6 (6p) tiene una prevalencia menor a uno por cada 100.000 nacidos vivos. Su fenotipo presenta una gran variabilidad dependiente del contenido génico de la región duplicada, e incluye bajo peso al nacer, retraso del desarrollo, anomalías craneofaciales, dificultades con la alimentación, infecciones respiratorias recurrentes, cardiopatías congénitas y anomalías renales5,6.
El objetivo es comunicar el caso de un paciente con diagnóstico por cariotipo de deleción parcial de 1p36.1 y trisomía parcial de cromosoma 6p.
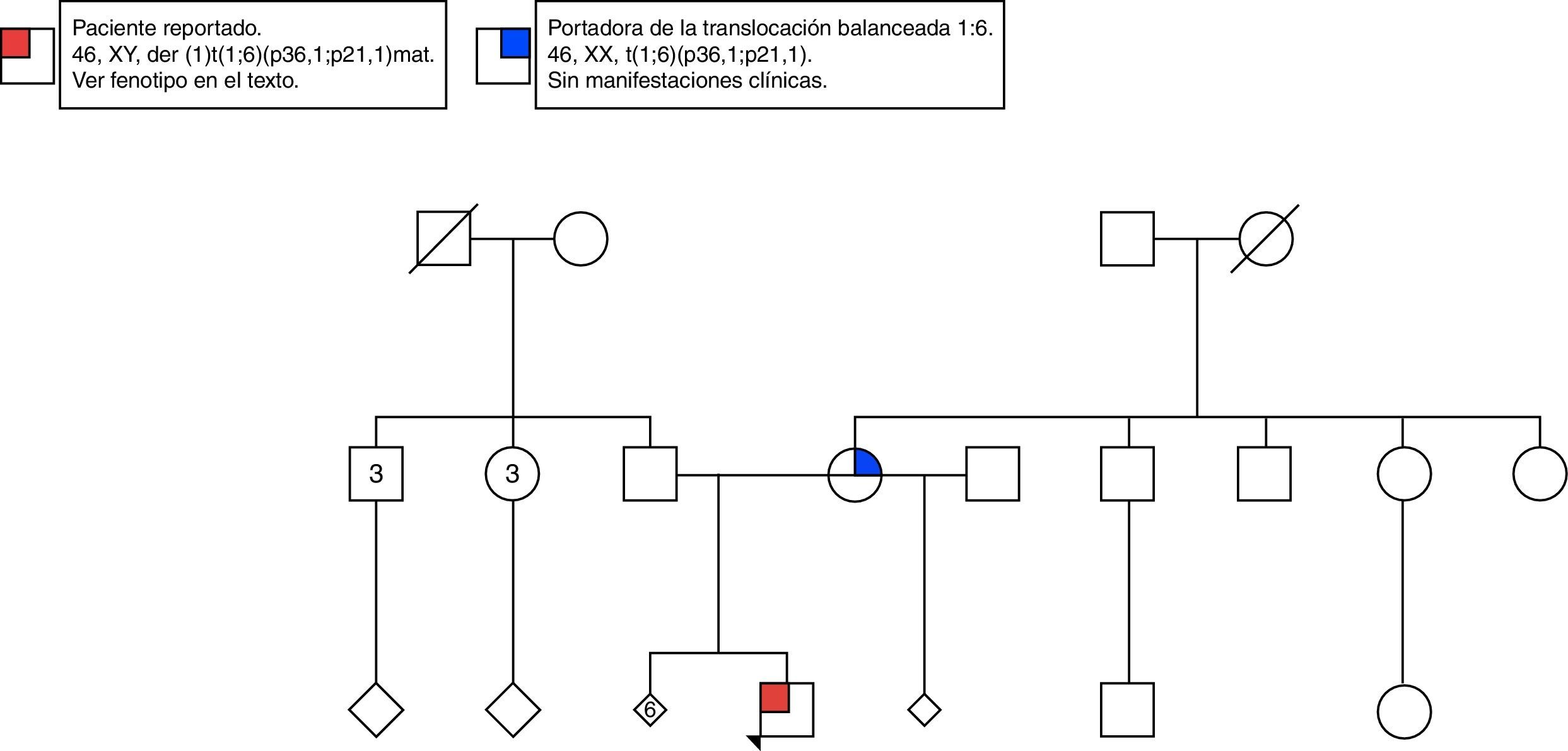
Caso clínicoLactante de 2 meses de edad, sexo masculino, parto atendido en un hospital nivel iii de la ciudad de Cali, en Colombia. Único hijo vivo de una mujer de 28 años con antecedente de abortos de repetición (fig. 1). Se le realizaron múltiples ecografías obstétricas que mostraron únicamente restricción del crecimiento intrauterino. Un doppler de la semana 36 evidenció vasodilatación cerebral aislada. Se realizó cesárea.

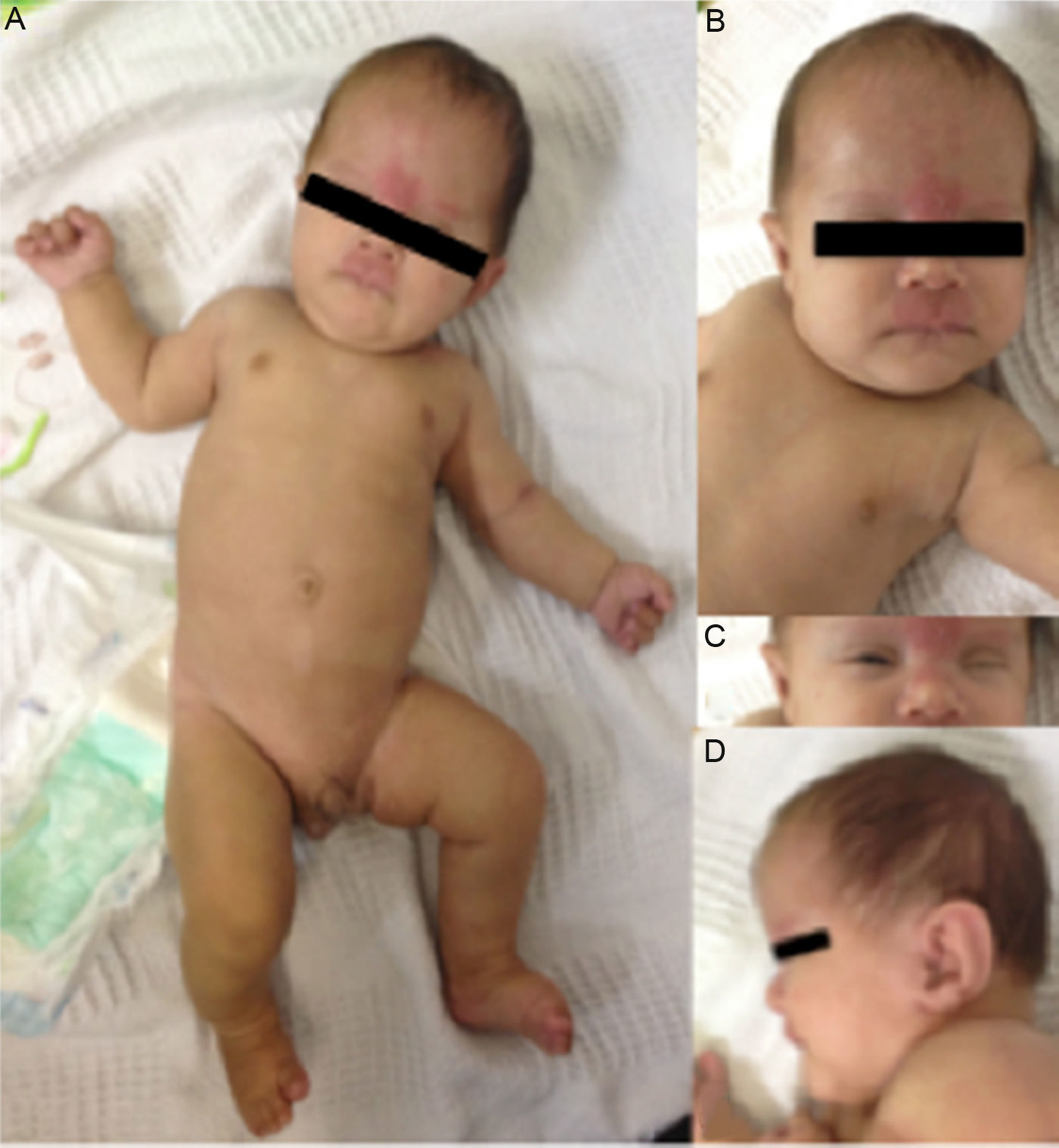
Al momento del nacimiento, con edad gestacional de 36 semanas, se encontró con un peso de 2.060g (percentil 4), talla de 48cm (percentil 34) y perímetro cefálico de 30cm (percentil 1). El lactante se hospitalizó en la unidad de cuidados intensivos neonatales por microcefalia y frente amplia. Posteriormente, el paciente se remitió a la consulta externa de dismorfología, a la cual llegó con 2 meses de edad y los siguientes hallazgos fenotípicos: una talla de 51cm (percentil 1), peso de 3.500g (percentil 1), un perímetro cefálico de 34cm (percentil 0), braquicefalia, fontanela anterior muy grande (7cm) que se continúa con la sutura metópica, frente amplia, cejas rectas, ojos hundidos, fisuras palpebrales cortas e inclinadas hacia arriba, epicanto, nevus flammeus en región frontal, nasal y labio superior, nariz corta, filtrum largo, labios delgados, frenillo lingual prominente, encía superior aserrada, mentón en punta, hipoplasia del tercio medio de la cara, orejas grandes con lóbulo pequeño y rotadas posteriormente, fositas preauriculares derechas, hemangioma en la nuca, fosita sacra, acortamiento rizomélico, pies y manos pequeños y criptorquidia unilateral izquierda. Además hipotonía y reflujo gastroesofágico (fig. 2).

A. Cejas rectas, acortamiento rizomélico, criptorquidia unilateral izquierda, pies y manos pequeños. B. Bosa frontal, nariz corta, filtrum largo, nevus flammeus en la región frontal, nasal y el labio superior, labios delgados. C. Ojos hundidos, fisuras palpebrales cortas e inclinadas hacia arriba, epicanto. D. Orejas grandes, con lóbulo pequeño y rotadas posteriormente; mentón en punta.
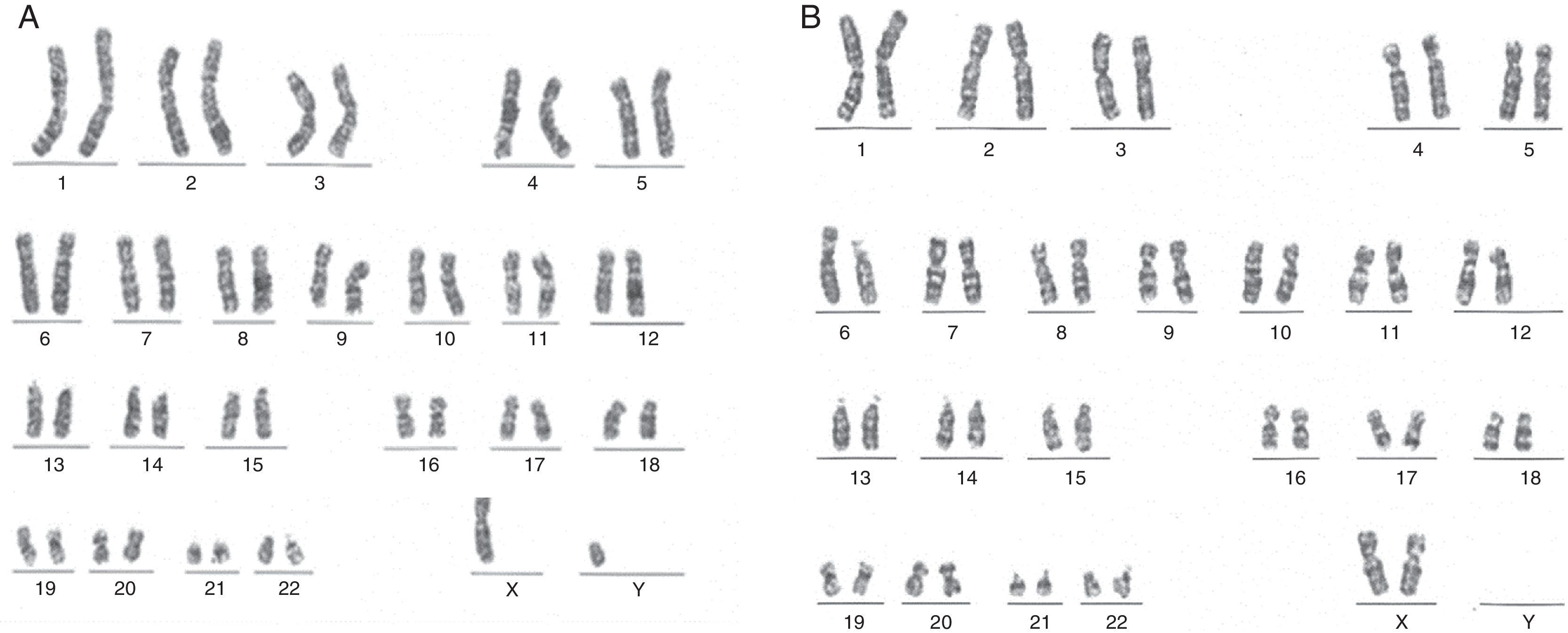
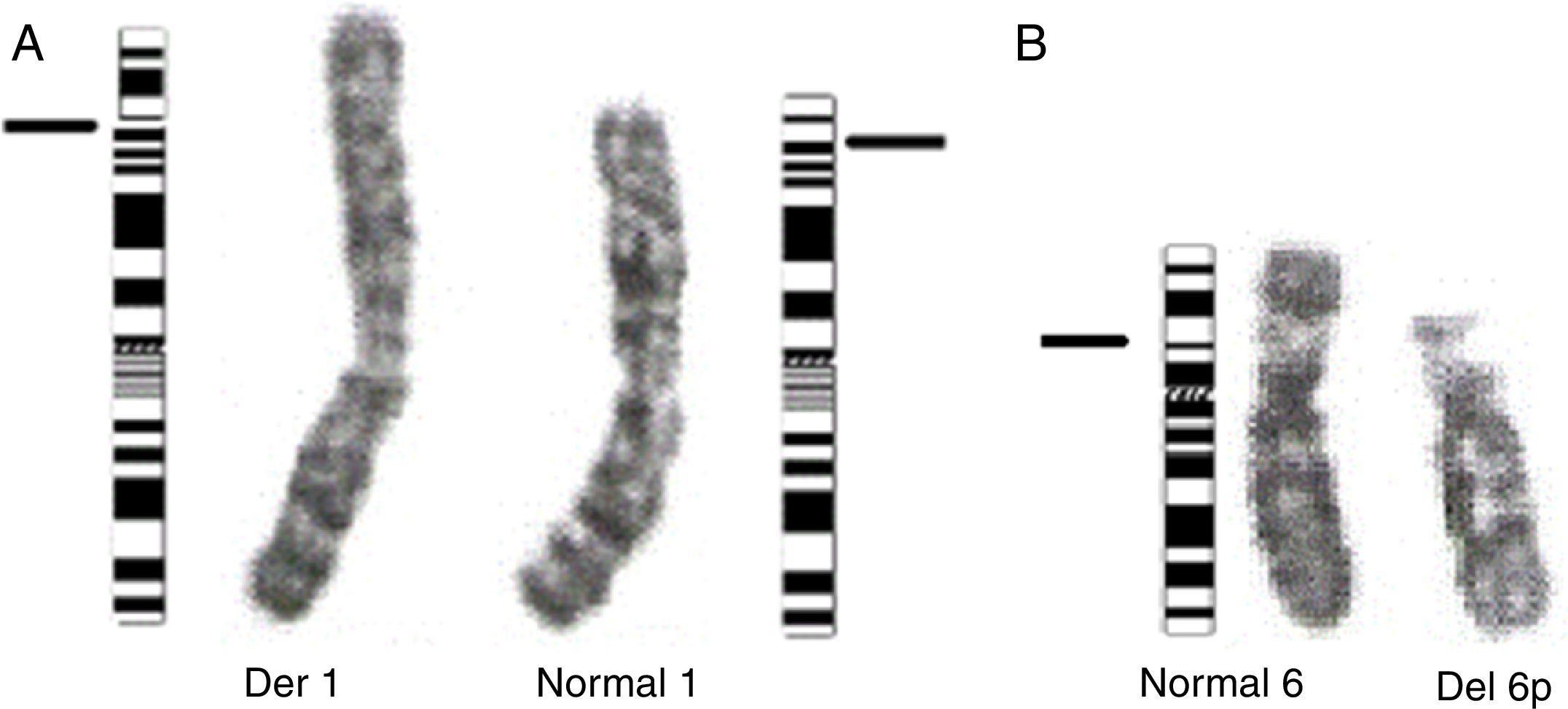
Se realizó cariotipo al paciente que reportó: 44,XY,der(1)t(1;6)(p36.1;p21.1) (fig. 3A). Se solicitó el cariotipo a los padres. El cariotipo de la madre reportó: 46,XX,t(1;6)(p36.1;p21.1) (fig. 3B). No fue posible realizar cariotipo al padre debido a que la madre no convivía con él y no fue posible contactarlo. Con estos resultados se evidenció que el paciente presentaba una alteración cromosómica estructural consistente en la presencia de un cromosoma 1 derivado de una translocación desbalanceada 1;6, con monosomía parcial del brazo corto del cromosoma 1 (p36.1 a pter) y trisomía parcial del brazo corto del cromosoma 6 (p21.1 a pter) (figs. 3A y 4A) de origen materno dado que la madre era portadora de una translocación balanceada 1;6 (figs. 3B y 4B).


Se dio asesoramiento genético a la madre, informando sobre los riesgos de tener hijos, alteraciones cromosómicas y aborto, informándole de que estos podrían ser tan altos como del 30% para cada evento, al ser portadora de la translocación; además se recomendó la realización de cariotipo a su madre (el padre está muerto), y según el resultado se definiría si es necesario plantear la realización de este análisis a sus hermanos.
DiscusiónLa deleción de 1p36 es la alteración cromosómica subtelomérica más frecuente, y tiene una incidencia estimada de uno por cada 5.000 nacidos vivos7. La presentación clínica es variable, e incluye características faciales como ojos hundidos, puente nasal deprimido e hipoplasia del tercio medio de la cara, así como un amplio rango de características clínicas no craneofaciales como cardiopatías, compromiso visual y/o auditivo, hipotonía, convulsiones, hipotiroidismo, pubertad precoz, obesidad, hiperfagia, trastornos de conducta, retraso global del desarrollo y discapacidad intelectual, entre otros2–4 (tabla 1). La mayoría de las veces las deleciones se originan de novo, sin embargo, un 3% de los casos se debe a segregación anómala de una translocación parental balanceada. El tamaño de la deleción varía desde submicroscópicas (<5Mb) a tan grandes como 32Mb8.
Principales características clínicas del síndrome de microdeleción 1p36 descritas en la literatura y presentes en el paciente reportado
| Características clínicas reportadas en el síndrome de microdeleción 1p36 y presentes en el caso reportado | Características clínicas reportadas en el síndrome de microdeleción 1p36 y ausentes en el caso reportado |
|---|---|
| Cejas rectas, filtrum largo, ojos hundidos, mentón en punta, hipoplasia mediofacial, fontanela anterior amplia con retraso de cierre, microcefalia, braquicefalia, orejas de implantación baja/posteriores/anormales, epicantus, fisuras palpebrales pequeñas, fisuras inclinación ascendente, pies/manos pequeños, anomalías esqueléticas (acortamiento rizomélico), anomalías gastrointestinales (reflujo), anomalías genitales externos (criptorquidia), hipotonía, dificultad para la alimentación, retraso del desarrollo, retraso del crecimiento | Puente/raíz nasal amplios, implantación baja del cabello, hipertelorismo, fisuras inclinación descendente, sinofridia, fisura palatina, braquidactilia/camptodactilia, clinodactilia, anomalías viscerales, miocardiopatía, defectos cardíacos congénitos, anomalías renales, anomalías EEG, anomalías cerebrales (RM/TC), convulsiones, disfagia orofaríngea, problemas auditivos, problemas oculares/visión, inatención visual, lenguaje pobre/ausente, RM, alteraciones del comportamiento, pubertad precoz, hipotiroidismo, obesidad |
Por otra parte, la trisomía distal de 6p es una anormalidad cromosómica que generalmente resulta de una mala segregación de una translocación familiar equilibrada, o una inversión pericéntrica, y se encuentra acompañada de otro desequilibrio cromosómico5. La región duplicada incluye casi siempre 6pter, aunque también se han descrito duplicaciones intersticiales de 6p con diferentes fenotipos dependiendo de su tamaño y localización9. El fenotipo consiste en retraso del desarrollo, retraso en el crecimiento pre y posnatal, dificultades para alimentarse, infecciones respiratorias recurrentes, defectos cardíacos congénitos, anomalías renales y un patrón de características craneofaciales específicas, entre las cuales las más frecuentes son las oculares. Dichas características craneofaciales son: microcefalia, frente prominente, hendiduras palpebrales pequeñas y cortas (blefarofimosis), ptosis del párpado superior, hipotelorismo ocular, cataratas, microcórnea, estrabismo, puente nasal prominente, nariz corta y bulbosa, boca pequeña con labios finos, mentón pequeño y en punta e implantación baja de las orejas con lóbulos poco desarrollados (tabla 2)5,6,10,11. Un listado de las características fenotípicas del caso, en comparación con lo reportado en la literatura para ambas cromosomopatías, se muestra en las tablas 1 y 2.
Signos característicos de trisomía parcial de 6p y presentes en el paciente reportado
| Características clínicas reportadas en la trisomía parcial 6p y presentes en el caso reportado | Características clínicas reportadas en la trisomía parcial 6p y ausentes en el caso reportado |
|---|---|
| Bajo peso al nacer, retraso del desarrollo, problemas para alimentarse, nariz corta, frente grande y prominente, occipicio plano, fontanela anterior amplia, nares pequeñas, boca pequeña o labios delgados, barbilla puntiaguda, hemangioma | Infecciones recurrentes, inmunodeficiencia, craneosinostosis, atresia coanal, defectos cardiacos, clinodactilia/sindactilia, anomalías en la pigmentación de la piel, anomalías renales (hipoplasia), preoteinuria, problemas de visión, córneas pequeñas, cataratas, estrabismo, caída del labio superior, blefaroptosis, blefarofimosis, filtrum largo o corto, hipertrofia gingival. Micrognatia, hipoacusia sensorineural, orejas de baja implantación, malformaciones auriculares |
En el caso aquí reportado el diagnóstico se hizo a través de cariotipo convencional. A pesar de que el cariotipo con bandas G tiene una resolución limitada en comparación con las nuevas técnicas de citogenética molecular12, las aneuploidías, que son ocasionadas por la ausencia o adición de un cromosoma, y las translocaciones, que son generadas por el desplazamiento de un segmento de un cromosoma a un nuevo lugar en el genoma, son frecuentemente detectadas por este método. Además, permite detectar deleciones o inserciones mayores a 10MB cuando el cariotipo tiene niveles de resolución entre 400 y 500 bandas, pudiendo mejorarse la detección al observar los cromosomas en prometafase a alteraciones superiores a 3-5MB13,14.
En casos de anormalidades cromosómicas numéricas, como una trisomía total o una monosomía completa, no existe indicación de cariotipo a los padres del sujeto, ya que usualmente son ocasionadas por errores que ocurren durante la gametogénesis15. En contraste, en casos de anomalías estructurales como deleciones o duplicación y translocaciones, siempre está indicado el cariotipo a los padres, para descartar la posibilidad de que uno de ellos sea portador de una translocación balanceada que se relacione con el fenotipo del paciente, como en el caso aquí reportado16. Además, cuando los productos (abortos, fetos muertos o nacidos vivos) tienen fórmulas cromosómicas con translocaciones balanceadas recíprocas, no recíprocas o robertsonianas, cromosomas en anillo e inversiones, también son indicaciones de cariotipo en los progenitores17.
Una translocación desbalanceada se puede producir cuando se hereda un cromosoma con material genético adicional o faltante de un progenitor que portaba una translocación balanceada. También puede ocurrir que la duplicación o deleción del material genético sea de novo y no heredada, los cariotipos de los padres sean normales y la alteración cromosómica en su descendientes haya ocurrido por un error en meiosis 1, dado un intercambio de material genético anómalo entre cromosomas no homólogos en la profase. En estos casos es poco probable que los padres tengan otro hijo con esta alteración17.
En diversos estudios se ha concluido que parejas en las que un integrante es portador de una translocación balanceada tienen un riesgo elevado de infertilidad, embarazos con resultados no satisfactorios como abortos, fetos muertos, muerte neonatal temprana y progenie, con anomalías cromosómicas estructurales y fenotipos secundarios17.
Para calcular riesgos específicos según la translocación en la pareja y las probabilidades se debe tomar en cuenta el estimado de tener gametos viables según el imbalance, la predicción del tipo de segregación para que haya gametos viables y el sexo del padre portador (es más frecuente de 3:1 en mujeres, ya que los hombres tienen más probabilidad de tener infertilidad)18. Aplicando la fórmula Stene y Stengel-Rutkowski para calcular el riesgo máximo de tener embarazos con anormalidades, se consideran el número de abortos y embarazos totales que han presentado los padres. El resultado se estima en 11% de presentar otro embarazo con anormalidades cromosómicas18.
ConclusiónEl diagnóstico de alteraciones cromosómicas estructurales, como en el caso aquí presentado, se puede realizar a través de cariotipo con bandas G, el cual detectó la monosomía parcial 1p36 y la trisomía parcial 6p en el infante, y una translocación recíproca en la madre. Esto sugiere que en países donde las técnicas de evaluación cromosómica molecular, como la hibridación genómica comparativa por microarreglos, no está disponible masivamente, el estudio citogenético convencional aún tiene vigencia para el diagnóstico y la asesoría genética en casos específicos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
