



Entre los linfomas de la zona gris (LZG) encontramos neoplasias con características compartidas entre un linfoma difuso de células B grandes (LDCBG) y un linfoma de Hodgkin clásico (LHC). Lo poco habitual de la patología combinado con la heterogenicidad de la enfermedad, su reciente descripción como entidad específica que conlleva a dificultad y reto diagnóstico, así como la falta de suficiente experiencia terapéutica hacen de la enfermedad una entidad compleja de difícil diagnóstico y reto terapéutico que justifica su descripción continua.
Se presenta una paciente con fiebre de un mes sin respuesta al manejo inicial, se estudió y realizó biopsia de ganglio inguinal izquierdo con resultado diagnóstico de LZG con características intermedias entre LDCBG y LHC. Aunque no existen guías establecidas para el manejo de esta entidad, la evidencia actual sugiere mejor respuesta en tratamientos dirigidos a LDCBG, misma terapia empleada en esta paciente con la cual se obtuvo respuesta favorable.
In the grey zone lymphomas (GZL), there are overlapping characteristics between diffuse large B-cell lymphoma (DLBCL) and classic Hodgkin lymphoma (CHL). The unusual nature of the pathology combined with the heterogeneity of the disease, its recent description as a specific entity, its diagnostic difficulty, and the lack of sufficient therapeutic experience justifies its continuous description.
The case is presented of a patient with a fever of one month onset, with no response to initial management. A left inguinal lymph node biopsy reported a diagnosis of GZL with intermediate characteristics between DLBCL and CHL. Although there are no established guidelines for the management of this condition, the current evidence suggests a better response in treatments meant for diffuse large B-cell lymphoma. This same therapy was used in this patient, with a favourable clinical outcome.
Entre los linfomas de la zona gris (LZG) encontramos aquellas neoplasias con características compartidas entre un linfoma difuso de células B grandes (LDCBG) y un linfoma de Hodgkin clásico (LHC), principalmente el linfoma de Hodgkin variedad esclerosis nodular (LHEN)1.
La presentación clínica general de estos pacientes está asociada a la manifestación de síntomas B (fiebre, diaforesis y pérdida de peso) presentes en 45-50% de los pacientes2,3, o a la presencia de adenopatías en estudio que pueden cursar asintomáticas. La edad media de presentación es de 35 años y predomina de forma general en hombres4.
Histológicamente se identifican características tanto de un LDCBG como de un LHC siendo común la presencia de grandes células pleomórficas en un fondo de estroma fibrótico difuso. Estas células tumorales pueden ser una reminiscencia tanto de LDCBG como de LHC en cuyo caso puede encontrarse la presencia de células de Reed Stemberg con sus variantes características. Se pueden identificar un amplio espectro de apariencias citológicas en diferentes áreas del tumor; el infiltrado inflamatorio suele ser escaso, pero pueden estar presentes eosinófilos dispersos, linfocitos e histiocitos5.
Inmunofenotípicamente presentan un patrón característico compartido de LDCBG y LHC, particularmente con asincronía entre morfología e inmunofenotipo. Pudiendo presentarse de esta forma con una morfología similar a LHC y un patrón fenotípico de LDCBG (CD20 ++, CD15-) o de forma inversa presentar una morfología similar a LDCBG con un fenotipo positivo para CD30, CD15 y pérdida de CD20 y CD79a. Los factores de transcripción PAX5, OCT-2 y BOB.1 son positivos en la mayoría de los casos.
A nivel genético se han identificado ganancias en regiones génicas por duplicación en los cromosomas 9p y 2p en variantes de ambos grupos de neoplasias6.
Respecto a la terapéutica no existe en este momento una guía de manejo establecida frente a esta entidad, en parte por lo poco habitual de la patología y en parte debido a la limitante de estudios enfocados con este objetivo, la evidencia reciente favorece el enfoque dirigido hacia LDCBG que ha mostrado mejores resultados en la sobrevida.
Caso clínicoMujer de 69 años que acude a consulta por fiebre de 38° de un mes de evolución. Contaba con un índice tabáquico de 40 sin otros antecedentes. Refirió la fiebre como insidiosa, de predominio vespertino y sin otros síntomas B. Recibió tratamiento antipirético y antinflamatorio sin mejoría; hace dos semanas se agregó dolor cólico en fosa renal izquierda irradiado a ingle ipsilateral por lo que se realizó ultrasonido donde se evidenció adenomegalia inguinal izquierda. Se realiza tomografía axial computarizada (TAC) que reportó la presencia de adenomegalias retroperitoneales paraaórticas de 3,3cm2 con extensión a región inguinal izquierda.
Acudió a nuestro hospital contando con signos vitales normales al ingreso, en la exploración física se identificó tumoración inguinal izquierda de 3cm2, indolora, indurada y no adherida a planos profundos. Los exámenes de laboratorio a su ingreso contaron con neutrofilia absoluta de 6771 en la biometría hemática; se solicitó un perfil bioquímico con normalidad. Reactantes de fase aguda elevados con PCR en 101,3 y VSG en 49mm/hr.
Se realizó linfadenectomía inguinal con toma de biopsia. Los hallazgos de inmunohistoquímica se encuentran en las figuras 1-3. Con estos hallazgos se realizó el diagnóstico de LZG y se inició tratamiento con esquema R-CHOP (rituximab más ciclofosfamida, doxorubicina, vincristina y prednisona) a dosis habitual con adecuada tolerancia planeándose ocho ciclos de quimioterapia; después del cuarto ciclo se realizó tomografía por emisión de positrones – tomografía computada (PET/TC) que no demostró actividad neoplásica; al completar el esquema se realizó un segundo PET/TC continuando con ausencia de la misma. Actualmente la paciente continúa con evolución favorable y se encuentra en seguimiento y vigilancia clínica sin evidencia de actividad de la enfermedad.
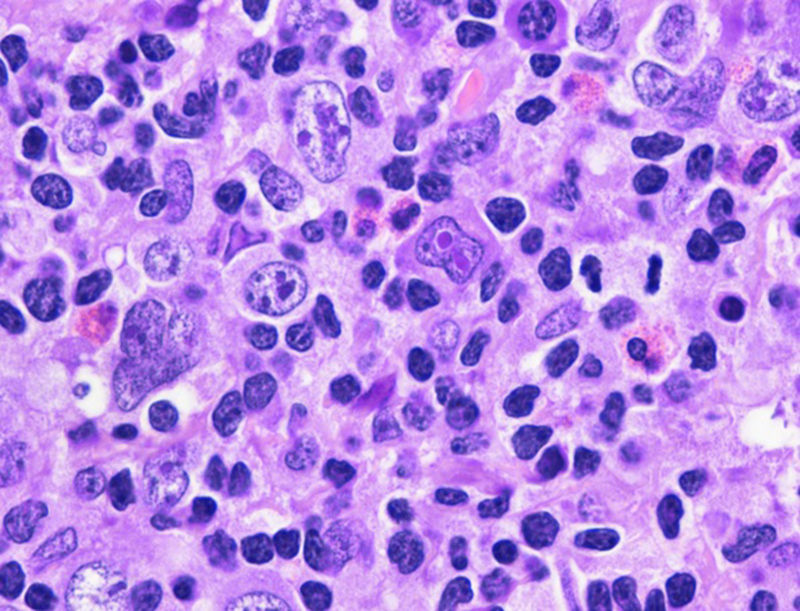
. Se observa pérdida de la arquitectura habitual con proliferación de abundantes células grandes de diverso tamaño y cantidad de núcleos variables, se identifica al centro una célula de Reed-Stemberg así como un fondo a base de polimorfonucleares, eosinófilos y linfocitos.")
Ganglio linfático (H-E 40x). Se observa pérdida de la arquitectura habitual con proliferación de abundantes células grandes de diverso tamaño y cantidad de núcleos variables, se identifica al centro una célula de Reed-Stemberg así como un fondo a base de polimorfonucleares, eosinófilos y linfocitos.


Los LZG son entidades neoplásicas poco frecuentes descritas inicialmente en pacientes con linfoma primario mediastinal de células B (LPMCB), un subtipo correspondiente a 10% de los LDCBG1. Aunque inicialmente se describió esta presentación con involucro mediastinal, estudios posteriores describieron una diferenciación entre la presentación mediastinal primaria: LZG con presentación mediastinal (LZGM) y presentación no mediastinal (LZGNM), aunque estos últimos cuentan hasta la fecha con relativa escasez de datos clínicos que les describan2,7. Existen otras entidades con solapamiento conocidas: LDCBG con linfoma de Burkitt, linfoma de Hodgkin con predominio nodular linfocitario (LHPNL) con LHC rico en linfocitos y LDCBG rico en células T e histiocitos con LHPNL8; entidades que de forma individual quedan fuera del propósito de esta revisión.
El término LZG se utilizó por primera vez en 1998 al describir pacientes con características intermedias entre LHC y otros subtipos de linfomas9. En 2005 se describieron formalmente los hallazgos patológicos de 20 casos de LZG diagnosticados y tratados por un período de 20 años, todos estos casos contando con afección mediastínica3.
Fueron reconocidos y clasificados por la Organización Mundial de la Salud a partir del año 2008 como un linfoma de células B no clasificable con características intermedias entre LDCBG y LHC, siendo una entidad relativamente nueva descrita principalmente como un LZGM debido a las primeras series de casos2,10. Esto se siguió de varios análisis histopatológicos, inmunofenotípicos, cromosómico y de alteraciones moleculares, así como de perfiles de metilación que identificaron diferenciación de esta entidad al presentar elementos compartidos entre LHC y LPMCB11,12.
Los LZGM son similares a LHC y LPMCB ocurriendo en adultos jóvenes entre la tercera y cuarta década de la vida y con incremento en la incidencia de enfermedad voluminosa (Bulky disease)13. Los LZGNM se presentan típicamente en personas de edad avanzada y en estadios tardíos de la enfermedad con mayor incidencia de involucro extranodal como ocurrió en nuestro caso2. Debido en parte a estas características, los pacientes con LZGNM cuentan con puntuaciones pronósticas incrementadas en comparación con pacientes con LZGM. A pesar de esto, los resultados finales no han demostrado diferencia significativa en la supervivencia, una posible explicación podría ser la mayor incidencia de enfermedad mediastinal masiva en pacientes con LZGM2. Los LZG derivan de una célula B alterada a nivel del timo; mismo precursor de LHC y LPMCB. Este origen común explicaría la posibilidad de recaída de alguna de estas tres neoplasias como alguna de estas mismas entidades y esclarece la presencia de neoplasias sincrónicas comunes13; como prueba de ello se ha demostrado que estas entidades comparten diversas anormalidades genéticas14.
El diagnóstico de LZG como ocurre en cualquier paciente con LDCBG o LHC, involucra la obtención de una biopsia del tejido afectado7,13. Puede encontrarse un amplio espectro de morfologías características tanto de LDCBG como LHC, se puede identificar también áreas morfológicas divergentes dentro de la misma lesión que requieren un muestreo extenso para un diagnóstico acertado13.
Las células neoplásicas suelen ser grandes y de aspecto centroblástico o inmunoblástico, pueden presentar alto grado de pleomorfismo e incluir células de Reed-Stemberg y sus variantes; también pueden estar presentes células momificadas. Los casos en que se asemejan a LPMCB las células neoplásicas se distribuyen en patrón de láminas de cebolla; en los casos que asemejan LHC las células neoplásicas son escasas y se esparcen en un fondo inflamatorio mixto con pequeños linfocitos, histiocitos y eosinófilos4. Los hallazgos en nuestra paciente se aprecian en la figura 1.
Estos casos pueden mostrar un grado significativo de fibrosis por lo que la distinción de un verdadero LHC resulta difícil. La necrosis puede estar presente y ser extensa. Los infiltrados de neutrófilos, sin embargo, no son una característica habitual4.
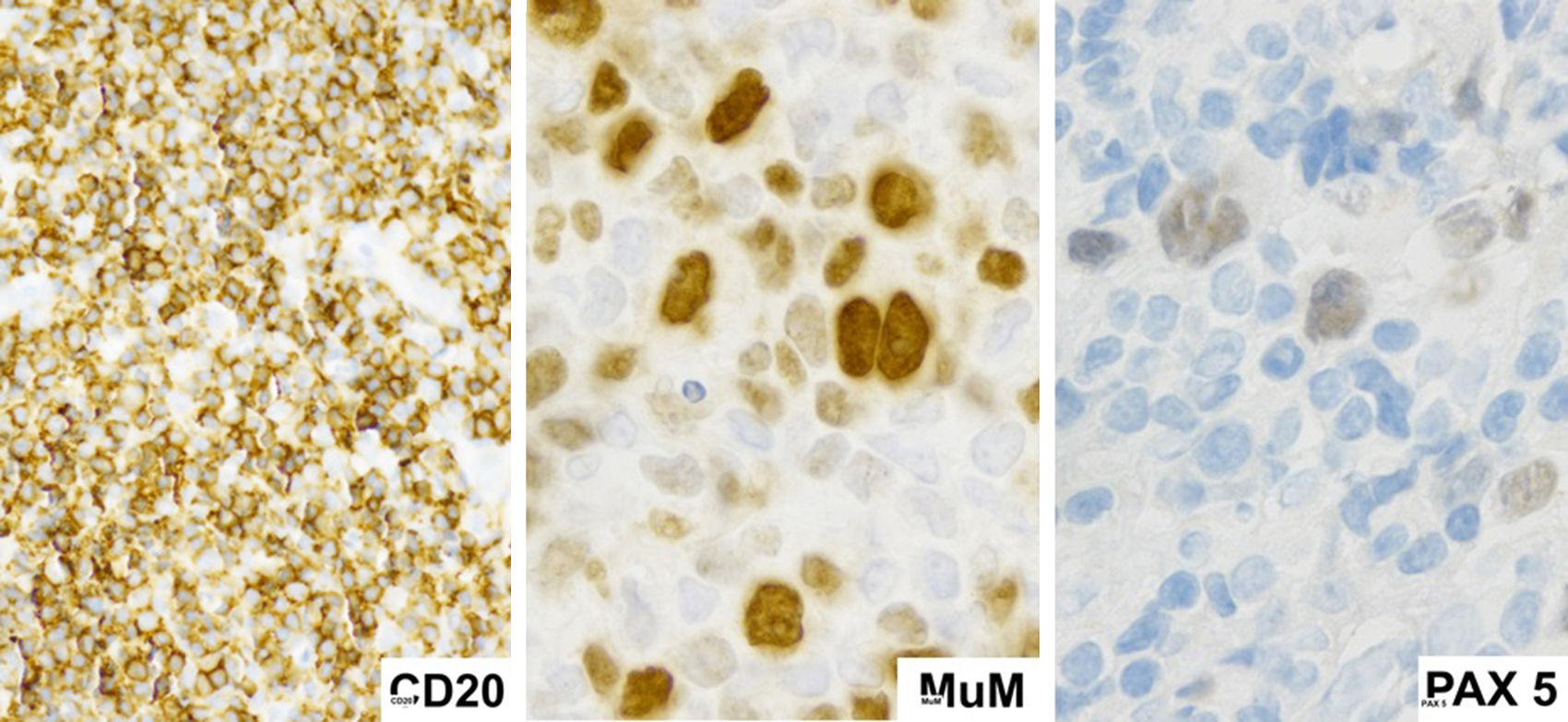
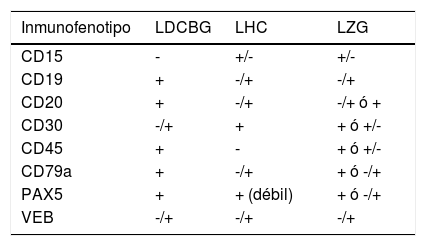
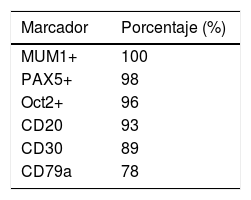
Al igual que ocurre con la morfología, el inmunofenotipo de LZG es variable y de características intermedias entre LCGB y LHC (tabla 1). Debido a que los factores de transcripción se encuentran preservados en estas células; PAX5, OCT2 y BOB1 habitualmente suelen encontrarse positivos13. Los inmunofenotipos de mayor prevalencia identificados se encuentran en la tabla 22.
Infunofenotipo característico: LDCBG, LHC, LZG
| Inmunofenotipo | LDCBG | LHC | LZG |
|---|---|---|---|
| CD15 | - | +/- | +/- |
| CD19 | + | -/+ | -/+ |
| CD20 | + | -/+ | -/+ ó + |
| CD30 | -/+ | + | + ó +/- |
| CD45 | + | - | + ó +/- |
| CD79a | + | -/+ | + ó -/+ |
| PAX5 | + | + (débil) | + ó -/+ |
| VEB | -/+ | -/+ | -/+ |
Abreviaciones: + todos los casos positivos, +/- la mayoría de los casos positivos, -/+ algunos casos positivos, - todos los casos negativos14,19.
En nuestro caso las células malignas expresaron CD15 y CD30, marcadores característicos de las células de Reed Stemberg de LHC, presentaron también expresión de CD19, CD20, CD45, CD79, y PAX5; característicos del linaje de células B, que es distinto al linfoma de Hodgkin en el cual la pérdida programada de estas es una característica biológica fundamental15.
En cuanto a análisis de inmunohistoquímica se ha identificado incremento de presencia en las tinciones para moléculas de muerte programada 1(PD-1) y sus ligandos (PD-L1 y PD-L2), obteniendo mayor presencia de PD-L1 en LHC y de PD-L2 en LPMCB. Los LZG presentan un patrón similar al de LHC con incremento en PD-L1 principalmente16.
Existen escasas recomendaciones respecto a enfoques terapéuticos en esta neoplasia en particular. En un estudio reciente de 112 pacientes con LZG tratados en 19 centros de Estados Unidos de los cuales 43% fueron LZGM y 57% LZGNM, los enfoques de tratamiento más utilizados fueron CHOP (ciclofosfamida, doxorubicina, vincristina, prednisona) en 51% de los pacientes y ABVD (doxorubicina, bleomicina, vinblastina, dacarbazina) en 30%, con un tercer grupo (10%) tratados con DA-EPOCH-R (etopósido, prednisona, vincristina, ciclofosfoamida, doxorubicina + rituximab)2. Aproximadamente dos tercios de los pacientes recibieron rituximab como parte de la terapia de primera línea en los tres grupos. Las tasas de respuesta global y las tasas de remisión completas para todos los pacientes fueron del 71% y 59% respectivamente; con 33% de pacientes con enfermedad refractaria primaria, sin presentar diferencias entre LZGM y LZGNM. Las tasas de respuesta global y las tasas de remisión completas de los pacientes que recibieron rituximab como parte del tratamiento de primera línea fueron 82% y 73%, respectivamente, frente a 59% y 43% sin rituximab. El tratamiento con un régimen de tipo LDCBG (es decir, DA-EPOCH-R o R-CHOP) pareció asociarse a una mayor tasa de sobrevida2.
En otro estudio que contó con 24 pacientes con LZGM tratados con DA-EPOCH-R, el 62% alcanzó remisión completa continua lo que demostró eficacia terapéutica17. En este mismo estudio los resultados se compararon con pacientes similares con diagnóstico de LPMCB. Tuvieron resultados significativamente inferiores en comparación con LPMCB: 5 años de supervivencia libre de eventos: 62% frente a 93% y supervivencia general de 5 años: 74% vs. 97% respectivamente, hecho que asocia un mayor grado de agresividad de esta neoplasia particular17,18.
Respecto a terapias biológicas, hay casos reportados de mejoría en pacientes con diagnóstico de LZG con el uso de brentuximab vedotin, anticuerpo conjugado contra CD30 con eficacia demostrada en LHCs refractarios, pero su eficacia en LZG no está aún definida20.
En concreto, los LZG son entidades con relativa descripción reciente, debido a la escasez actual de criterios clínicos, histopatológicos e inmunohistoquímicos se justifica la descripción continua de la entidad para su identificación oportuna, y de esta manera disminuir su retraso diagnóstico que conlleve a un abordaje terapéutico temprano que condicione la mejoría en los resultados clínicos y de remisión final en los pacientes.
No existe en este momento una guía de manejo establecida frente a esta entidad, en parte por lo poco habitual de la patología cuyos diagnósticos diferenciales principales son un LDCBG por un lado o un LHC por el otro. Este hecho es de vital importancia debido a que ambas neoplasias encaminan a abordajes terapéuticos y resultados finales distintos; uno de los puntos clave radica en la confusión por considerar únicamente alguna de estas 2 entidades como posibilidad diagnóstica aislada, dejando de lado una tercera opción con pronóstico y abordaje terapéutico distintos: un LZG. Fuera de los resultados por inmunohistoquímica necesarios para el diagnóstico, en nuestra paciente no se realizaron pruebas genéticas mismas que son de utilidad para confirmar las características alteradas de estas neoplasias y corroborar el diagnóstico.
Existe la limitante de estudios dirigidos a la terapéutica; los enfoques actuales parecen evidenciar una mejor respuesta y tasa de supervivencia en tratamientos dirigidos hacia LDCBG como ocurrió con nuestra paciente, la cual se encuentra actualmente en remisión tras una respuesta favorable al tratamiento con esquema R-CHOP encontrándose actualmente asintomática, sin presencia de adenopatías clínicas identificables y con reducción de las mismas por imagen; sin embargo, aún no se cuenta con suficiente evidencia que respalde por completo este enfoque, hecho que justifica la consideración de otros posibles abordajes y esquemas terapéuticos, así como el continuo seguimiento de estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
