


El astrocitoma con diferenciación gemistocítica es una variante histológica de los astrocitomas difusos que se caracteriza por presentar tendencia a una rápida progresión hacia la malignidad, y a pesar de ser clasificado según la Organización Mundial de la Salud como un glioma grado II, presenta pobre pronóstico a corto plazo. La presentación clínica más común de este tipo de tumores son los síndromes convulsivos y los déficits focales propios de la localización, mientras que el síndrome de hipertensión endocraneana es poco común. Como primera manifestación se encuentra irritación meníngea más aún en ausencia de infección del sistema nervioso central. Se reporta el caso de un paciente que ingresó a servicio de urgencias con irritación meníngea, a quien finalmente se le diagnostica astrocitoma gemistocitico intraventricular.
An astrocytoma with gemistocytic differentiation is a histological variant of diffuse astrocytomas, characterised by a tendency towards a rapid progression to malignancy. Despite being classified as a grade II glioma according to the World Health Organisation, it has poor short-term prognosis. The most common clinical presentation of these types of tumours are, convulsive syndrome, focal deficits secondary to the location of the tumour, and / or intracranial hypertension syndrome. It is uncommon to find meningeal irritation as a first manifestation, and even more, in the absence of infection of the central nervous system. The case is presented on a patient, who was admitted to the Emergency Department with signs of meningeal irritation. A diagnosis of intraventricular gemistocytic astrocytoma was finally made.
El astrocitoma gemistocítico (GemAs) es una variante histológica de los astrocitomas difusos, que se caracteriza por presentar astrocitos neoplásicos con una gran expresión de proteína ácida fibrilar glial (PAFG) y tendencia a una rápida progresión hacia la malignidad1,2. Se presenta caso de paciente con diagnóstico histopatológico e inmunohistoquímico de astrocitoma con diferenciación gemistocítica, quien presentó cuadro de síndrome meníngeo como primera manifestación de GemAs.
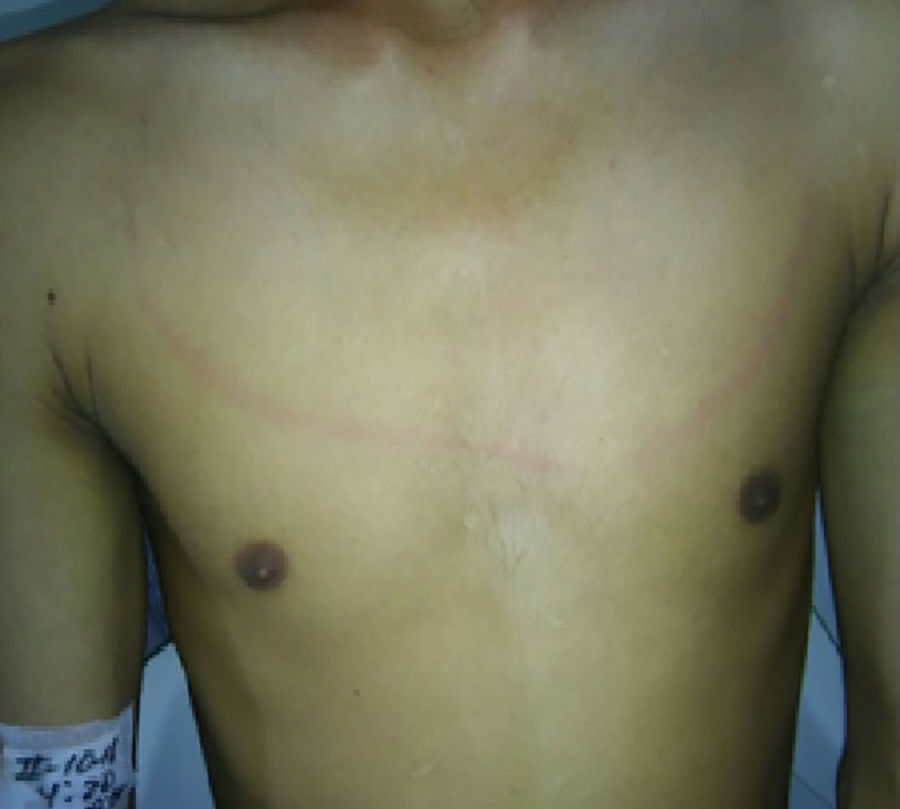
Caso clínicoPaciente masculino de 21 años quien consultó por cuadro clínico de 4 días de duración, el cual inició con cefalea intensa en región occipital asociado a mareo y vómito. El paciente además refiere que desde hace 10 días presenta episodios de disfagia a los sólidos, los cuales empeoraron con el inicio de la cefalea. En el examen físico el paciente se encontraba afebril, alerta y orientado en las tres esferas mentales, con lenguaje apropiado, pupilas isocóricas, fotorreactivas sin déficit sensomotor, no presentaba signos cerebelosos ni signos meníngeos clásicos Brudsinski ni Kering, pero presentaba rigidez nucal y una evidente raya meníngea en la pared torácica anterior al estímulo sensitivo (fig. 1). Los paraclínicos de ingreso descartan signos de síndrome de respuesta inflamatoria sistémica (SIRS), hemograma normal con PCR negativa. El paciente refiere haberse realizado esquemas de vacunación y niega otro tipo de antecedentes (fig. 1).

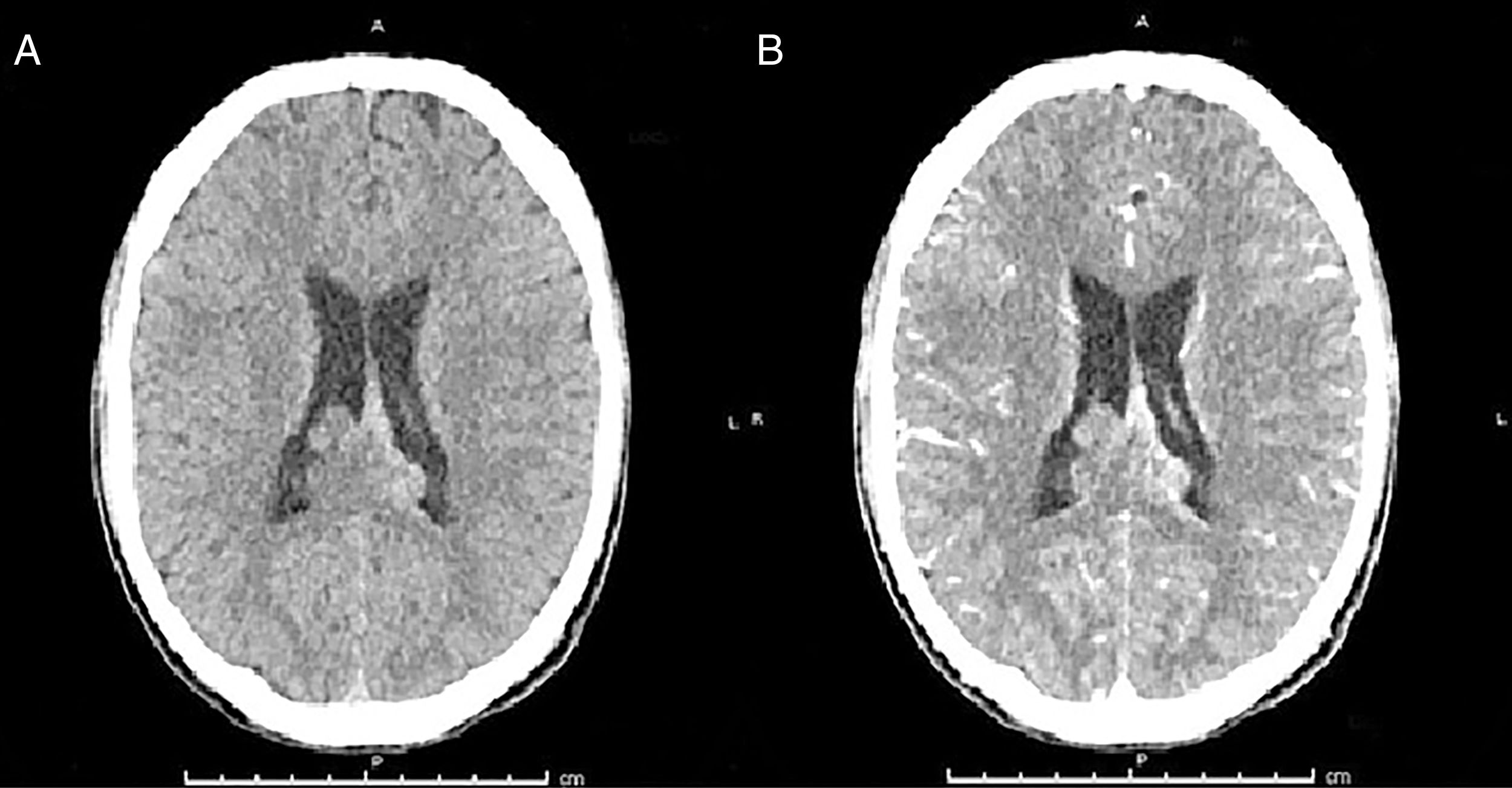
Por la sintomatología descrita, se plantea el diagnóstico de síndrome meníngeo solicitándose tomografía axial computarizada (TAC) cerebral simple y contrastado, y punción lumbar (PL). En el estudio imagenológico se observó lesión de carácter sólido supratentorial parietal que traspasa la línea media, que condiciona la ligera dilatación del sistema ventricular y de probable origen neoplásico (fig. 2). La punción lumbar reporta líquido cristalino con presión de apertura elevada, su estudio citoquímico inicial mostró leucocitos 3 por milímetro lactato 6,2 mmol/dl, hiperproteinorraquia 69mg/dl, glucorraquia de 10mg/dl, glicemia de 125mg/dl. Se le ordena al paciente protocolo antibiótico bajo la sospecha de meningitis y administración de esteroides, además manejo de analgésico (fig. 2).

Tomografía cerebral, cortes axiales: A. TAC cerebral simple B. TAC cerebral contrastada. Se observa lesión expansiva que compromete el cuerpo calloso, septum pellucidum en zona posterior, con extensión al epéndimo y cuerpos ventriculares con realcenodular en las lesiones interventriculares, bilaterales.
Se realizó imagen por resonancia magnética (IRM) contrastada para aclarar los hallazgos obtenidos en el TAC. La IRM reporta lesión que compromete los ventrículos laterales y que infiltra cuerpo calloso con poco realce, con el medio de contraste sugestivo de ependimoma; por lo anterior, se ordena realización de biopsia endoscópica intraventricular, con reportes de Gram y cultivo de LCR negativo, estudio con tinción de Zielh-Nielsen negativo, tinción de tinta china en LCR negativo, y cultivo para hongos de LCR, se suspende administración de antibioticoterapia.
En el acto quirúrgico, se lleva a cabo la craneotomía para drenaje y extracción de tumor intraventricular, realizándose desperiostización con trepanación, incisión dural en cruz. Posteriormente se avanza el endoscopio hacia el ventrículo lateral derecho y se navega por el cuerpo del ventrículo hasta visualizar la lesión pediculada, adherida al techo del ventrículo lateral. Se realiza resección subtotal en piezas, lavado y hemostasia, retiro del endoscopio y cierre de planos. En el postquirúrgico el paciente ingresa de inmediato a Unidad de Cuidados Intensivos despierto, extubado, estable hemodinámicamente y sin déficit sensitivo o motor. TAC cerebral de control, reporta cambios residuales postquirúrgicos y persistencia de la lesión interventricular.
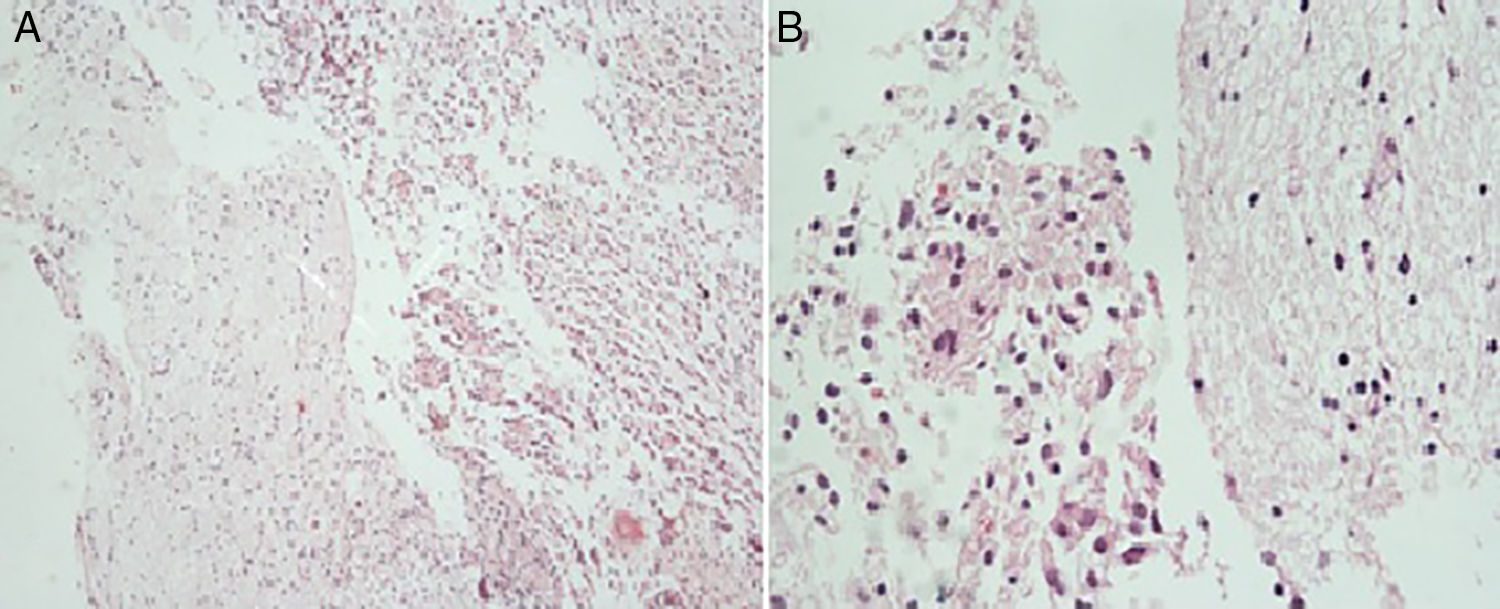
El estudio histopatológico informa astrocitoma con diferenciación gemistocítica (WHOII). Se realizaron estudios de inmunohistoquímica que demuestran reactividad difusa para p53 y proteína gliofibrilar ácida (GFAP) con reactividad de células acompañantes para WT1 y neurofilamentos. Se reporta una proliferación celular determinada con índice de Ki67 de un 20%.
Posteriormente el paciente continúa con cefalea y emesis pese al tratamiento médico, evoluciona desfavorablemente, presentado cefalea intensa, urente, holocraneana tornándose confuso. Desarrolla parálisis facial periférica izquierda y voz gangosa por parálisis de pares craneales bajos. En los días siguientes presentó anisocoria dada por midriasis derecha y hemiparesia izquierda. El paciente desarrolla alteraciones hemodinámicas y metabólicas que finalmente lo llevan al fallecimiento a los 15 días del procedimiento quirúrgico.
DiscusiónEn 1935, Elvidge et al. introdujeron el término astrocitoma gemistocítico para definir un subtipo de astrocitoma caracterizado por la presencia de ̈un cultivo casi puro de gemistocitos̈ (astrocitosgemistocíticos)3. El término gemistocito, proveniente de la palabra griega gemistos significa ̈llenö, haciendo referencia a su tamaño (15 – 40μm), células ovaladas o redondas con abundante citoplasma4. Fue descrita por primera vez como “Glía inflamada” en los inicios del siglo XX por Franz Nissl, el cual interpretó este tipo celular como una variante de astrocitos asociada a estados patológicos5,6.
La actual clasificación de la Organización Mundial de la Salud (OMS) define al GemA como una variante de los astrocitomas difusos, caracterizada por la presencia de al menos 20% de astrocitosgemistocíticos neoplásicos. Aun así, el porcentaje de gemistocitos definido para realizar el diagnóstico es arbitrario y basado en datos muy limitados6,7.
Esta neoplasia se caracteriza histopatológicamente por la presencia de gemistocitos, células tumefactas de citoplasma eosinófilo brillante e hialino, del que surgen abundantes prolongaciones robustas, tipo celular que también puede encontrarse en el glioblastoma2,8–12. La presencia de núcleo excéntrico con cromatina densamente condensada e infiltración linfocítica perivascular también puede ocurrir asiduamente, como también la formación de cúmulos celulares2,6,7. Es poco frecuente encontrar la forma pura de esta clase de neoplasia, ya que es más usual que los tumores cumplan con los criterios de grado III de la OMS10. Pese a eso, no se catalogan como gliomas de alto grado porque no hay suficiente evidencia que refiera que la morfología gemistocítica per se sea factor independiente de mal pronóstico6. Las características histológicas de la masa resecada al paciente se muestran en la figura 3.

El origen y el significado de este tipo celular no son claros. Hoshino et al. han reportado que los gemistocitos no sintetizan ácido desoxiribonucléico (ADN), pero ocasionalmente incorporan tritiada de timidina en sus estados originales como astrocito fibrilar o protoplásmico, induciendo una transformación hacia astrocito gemistocítico iniciando su ciclo de proliferación. De acuerdo con estos autores los gemistocitos reflejan la proliferación de las células adyacentes a estas, pero los gemistocitos propiamente son células inertes. No obstante, esta hipótesis no explica la presencia de tumores agresivos con una población predominante de gemistocitos4,6,7,11.
Esta variante de astrocitoma con diferenciación gemistocítica representa de 9% - 19% de los astrocitomas. Algunas series reportan la presencia de esta patología en poblaciones con edades medias de 38,2 años hasta 42,4 años3,6,7. Pese a ser considerados como astrocitomas grado 2, estos tumores presentan una alta propensión a transformación maligna7,9,12. Babu et al. reportaron que el GemAs presenta una peor sobrevida al ser comparado con otros subtipos, ya que los pacientes que presentaron la variante gemistocítica sobrevivieron menos de la mitad de la supervivencia media que presentaron aquellos pacientes con astrocitoma fibrilar y protoplásmico2.
Macroscópicamente se reconocen como tejidos grisáceos, oscuros, granulares con apariencia quística variable y los bordes son de difícil limitación en las piezas quirúrgicas, estas características también pueden ser encontradas en neoplasias infiltrantes6. Estos tumores son bien diferenciados de crecimiento lento, pero muestran una tendencia constante al infiltrar difusamente el tejido cerebral adyacente. Por esta razón pueden progresar hacia astrocitomas anaplásicos o glioblastomas, lo cual es concordante con lo sucedido al paciente. Rusell y Rubinstein reportaron una conversión hacia glioblastoma en el 80% de los casos de GemAs analizados12. Esta tendencia hacia la progresión es característica de este subtipo de gliomas de bajo grado, lesiones en las que encontramos una gran expresión de PAFG, la cual forma una red fibrilar2,13. La explicación de esta tendencia a la progresión maligna no es clara, ya que los gemistocitos propiamente tienen una baja tasa de proliferación que es determinada por timidinatritada, bromodeoxyuridina y anticuerpos monoclonales Ki-/MIB-11,7,14.
En el estudio realizado por Krouwer et al., catalogaron como GemAs puro aquel que presentaba una población de gemistocitos mayor a 60% por campo de alto poder (CAP) y como mixto el que presentaba de 20% a 60% de gemistocitos por CAP7. En este mismo estudio encontraron que la media de edad fue significativamente mayor en el grupo de GemAs puros respecto a la de mixtos, 48,5 vs. 38,3 años; < 0,05, lo que sugiere que la edad menor a 50 años en los pacientes con GemAs tendría una influencia en un mejor pronóstico al igual que la duración de la sintomatología preoperatoria menor a 6 meses y la ausencia de convulsiones como síntoma inicial. Sin embargo, la mayoría de pacientes en ambos grupos reportaron haber tenido como primera manifestación convulsiones y duración de síntomas preoperatorios de 21,9 para ambos grupos7. Esto contrastado con lo hallado por Yang et al., quienes encontraron que no había diferencia significativa entre estos dos grupos en estos aspectos14.
Los astrocitomas gemistocíticos muestran una gran variedad de alteraciones genéticas, en su estudio Watanabe K et al. encontraron que el 83% de los astrocitomas gemistocíticos analizados presentaban mutación de p53, y de estos un 91% eran mutaciones asociadas a errores en puntos de codificación. Otro aspecto importante fue que el 100% de los astrocitomas de bajo grado que progresaron a malignidad presentaron p53(+) acompañado de una tasa de gemistocitos mayor al 5%, mientras que la mutación PTEN se encontró asociada a un subgrupo de astrocitomas anaplásicos mas no de astrocitomas gemistocíticos.
También encontraron que la característica histológica prominente era la presencia de células neoplásicas gemistocíticas con un índice 35,0+- 9,9 con un rango entre el 15–58%, este valor coincide con el propuesto en el estudio realizado por Krouwer, el cual propone un valor de corte de mínimo 20% para esta variante, punto de corte que además tiene relación con la sobrevida a 5 años1,7. En la revisión realizada se identificó que la presentación clínica más común del GemAs, son: episodios convulsivos, déficits focales dependientes de la localización de la lesión, cefalea e infrecuentemente síndromes de hipertensión endocraneana15. En esta misma revisión no se identificó al síndrome de irritación meníngea como manifestación primaria del GemAS, más aún sin criterios completos de meningitis infecciosa como es el caso de nuestro paciente. Se ha establecido que las manifestaciones clínicas primarias de síndromes de irritación meníngea, en presencia de masas intracraneales, son en su mayoría relacionadas con la presencia de tumores pituitarios, ya sea por la creación de una fístula de LCR a través de la lámina cribosa o apoplejía pituitaria. Esta última ocasionando que el tejido necrótico penetre al espacio subaracnoideo, causando así hipoglucorraquia pleisotosicis e hiperproteinoraquia16,17. Sin embargo, ninguna de estas dos condiciones estuvo presente en el paciente, por lo que podría contemplarse que el cuadro de síndrome de irritación meníngea y los hallazgos en el LCR son propios de la ubicación del GemAs, que en este caso se localizaba en el techo del sistema ventricular, hecho que también explicaría los niveles aumentados de lactato.
Finalmente, concluimos que la evidencia existente ha permitido clarificar algunos aspectos sobre el astrocitoma con diferenciación gemistocítica, aun así algunas preguntas como el mecanismo por el cual se induce la formación de gemistocitos y si esta morfología es un factor independiente de pobre pronostico clínico siguen sin recibir respuesta definitiva. Por lo tanto, es necesario ampliar la información al respecto y tener un enfoque claro en el diagnóstico histopatológico e inmunohistoquímico de este tipo de neoplasias. De igual manera su presentación mediante el síndrome de irritación meníngea es infrecuente, dadas sus características ya explicadas y pobre pronóstico debería ser tenido en cuenta más seguido dentro de los diagnósticos diferenciales.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
