



La detección temprana del cordoma constituye un desafío médico dada su baja frecuencia, calculada entre 2-4% de todos los tumores primarios del hueso, requiriendo un alto índice de sospecha clínica. El diagnóstico tardío incrementa la morbimortalidad y compromete la sobrevida del paciente, por lo que el conocimiento actualizado sobre su etiopatogenia, clínica y tratamiento es de suma importancia para el cirujano espinal, quien forma parte esencial del grupo interdisciplinario terapéutico.
Reporte de casosReportamos dos casos de cordomas cervicales, localmente agresivos sin extensión sistémica, el primero de ellos ubicado en la columna cervical y el segundo en la unión craneocervical. Ambos con diferentes formas de presentación, síntomas, tiempo de evolución (5 años y 7 meses respectivamente), infiltración tumoral y abordaje quirúrgico, pero similar tratamiento oncológico. El objetivo del manuscrito es demostrar la variedad de presentación cervical de la entidad y las diferentes formas de abordarla. Se incluye una revisión actualizada de la literatura donde se evidencia la importancia de la inmunoterapia como nuevo horizonte terapéutico, y se reafirma la resección quirúrgica como base del mismo.
DiscusiónSe presentan los resultados imagenológicos de descompresión medular, resección ósea y de partes blandas, los tipos de fijación espinal y la evolución clínica, sin recidiva local a los 2 años y a los 12 meses respectivamente en cada caso, luego del manejo quirúrgico y la radioterapia adyuvante.
Nivel de evidencia: IV
Early detection of chordoma is a medical challenge given its low frequency, a high index of clinical suspicion is required for the diagnosis. The late diagnosis increases morbidity and mortality and compromises the survival of the patient. Knowledge about its etiology, clinical manifestations and treatment is of utmost importance for the spinal surgeon as well as for the therapeutic group. The aim of the report is to demonstrate the variety of cervical presentation of the entity and the different ways of approaching it.
Case ReportWe report two cases of locally aggressive cervical chordomas without systemic extension, the first one located in the cervical spine and the second in the craniocervical junction. Both with different forms of presentation, symptoms, evolution time (5 years and 7 months respectively), tumor infiltration and surgical approach, but similar oncological treatment.
DiscussionThe imaging results of medullar decompression, bone and soft tissue resection, types of spinal fixation and clinical evolution were presented. No local recurrence at 2 years and 12 months respectively after surgical management and adjuvant radiation therapy were found.
Evidence Level: IV
El cordoma representa un infrecuente tumor maligno primario del hueso, derivado de remanentes anormales de la notocorda en el esqueleto axial, característicamente de crecimiento lento, pero altamente agresivo por su capacidad de crecimiento local1.
Representa entre el 1% y el 4% de la totalidad de los tumores óseos primarios y el 0.5% de las neoplasias intracraneales, siendo el tumor primario maligno más frecuente de la columna cervical del adulto, luego de las lesiones linfoproliferativas, con una incidencia reportada entre 0.1/100.000 en hombres y 0.06/100.000 en mujeres2.
Debido a su baja frecuencia, la dificultad para realizar investigaciones clínicas y la inefectividad de la quimioterapia sistémica, el tratamiento de elección continúa siendo la resección quirúrgica en bloque y la radioterapia (RT) postoperatoria, con la significativa morbilidad asociada a corto plazo. A pesar de una resección agresiva, las recurrencias tumorales son comunes3.
Las nuevas técnicas de RT como las soportadas en iones de carbono y protones, reportan resultados prometedores para el manejo de cordomas primarios, estando ampliamente disponibles, facilitando el uso de altas dosis locales y haciéndolas modalidades terapéuticas seguras4.
Previa autorización escrita y firmada por los pacientes, presentamos dos casos de cordoma cervical con diferente grado de invasión, destrucción tisular y crecimiento tumoral, manejados mediante resección quirúrgica y RT. Realizamos una revisión de la literatura actualmente disponible.
Reporte de casosCaso 1Mujer de 49 años de edad, con historia médica de HTA controlada. Presentación inicial de cervicalgia progresiva de 5 años de evolución, hasta mantener un dolor en EVA de 8/10 en el momento de la valoración, asociada en los últimos 9 meses a disfagia para sólidos, que empeoró 2 meses antes también para líquidos. Valorada 2 años atrás de manera extrainstitucional con hallazgo de lesión tumoral en columna cervical, sin referir etiología, con indicación de manejo quirúrgico rechazado por la paciente.
A la exploración física, ausencia de compromiso general, hallazgos de contractura muscular paravertebral cervical dolorosa con limitación para los ROM en extensión, rotación e inclinación lateral de la cabeza, disestesia en cara lateral izquierda de cuello, sin déficit neurológico ni signos de mielopatía.
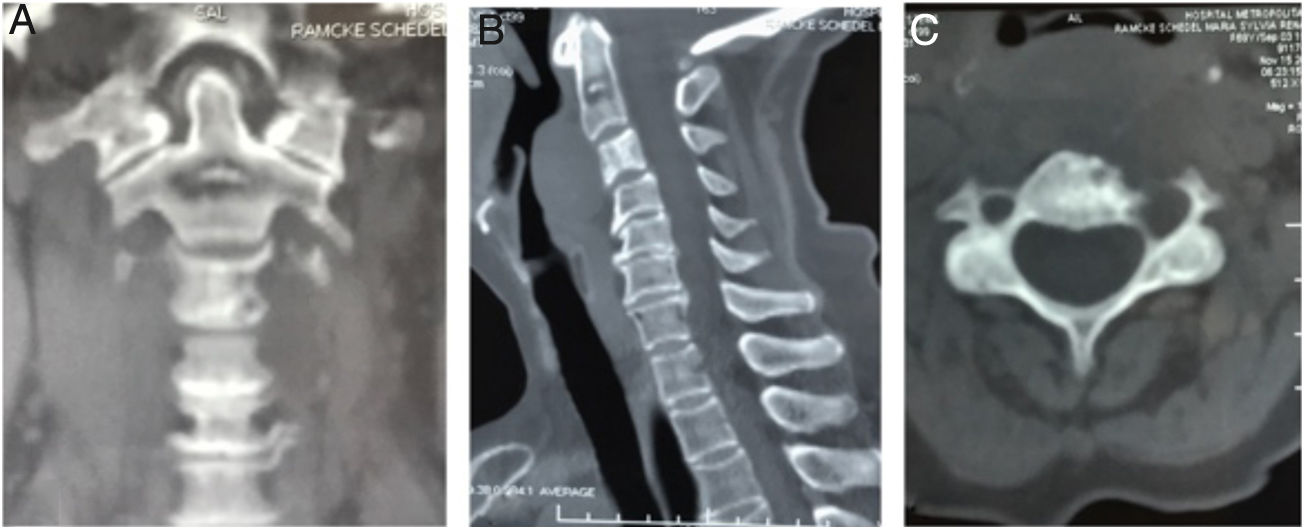
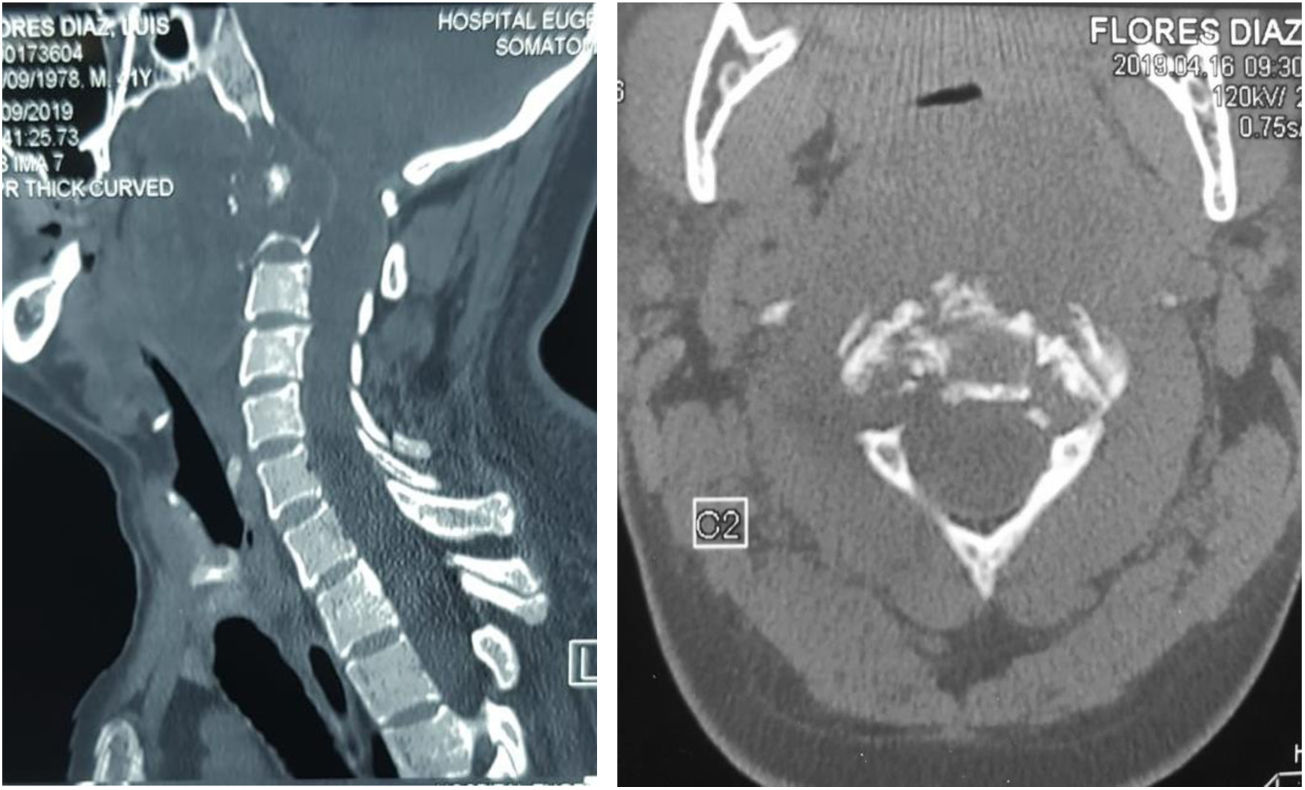
Pruebas complementarias negativas para infección, Calcio y Fósforo séricos dentro de parámetros de normalidad. Las imágenes de TAC (fig. 1) y RMN (fig. 2) mostraron una lesión ósea a nivel del cuerpo de C3, con masa tumoral prevertebral desde C2 hasta en nivel discal C3-C4.
. Corte coronal que muestra imagen lítica en el cuerpo de C2 (Lesión satélite). B) Corte sagital con lesión lítica a nivel de la odontoides y calcificación intralesional. Cambios artrósicos C4-C7. C) Corte axial de C3 donde se aprecia lesión lítica en cortical anterolateral izquierda de cuerpo vertebral, con zona esclerótica (lesión marginal) con aumento del diámetro de agujero transverso ipsilateral.")
Tomografía axial computarizada columna cervical. A). Corte coronal que muestra imagen lítica en el cuerpo de C2 (Lesión satélite). B) Corte sagital con lesión lítica a nivel de la odontoides y calcificación intralesional. Cambios artrósicos C4-C7. C) Corte axial de C3 donde se aprecia lesión lítica en cortical anterolateral izquierda de cuerpo vertebral, con zona esclerótica (lesión marginal) con aumento del diámetro de agujero transverso ipsilateral.
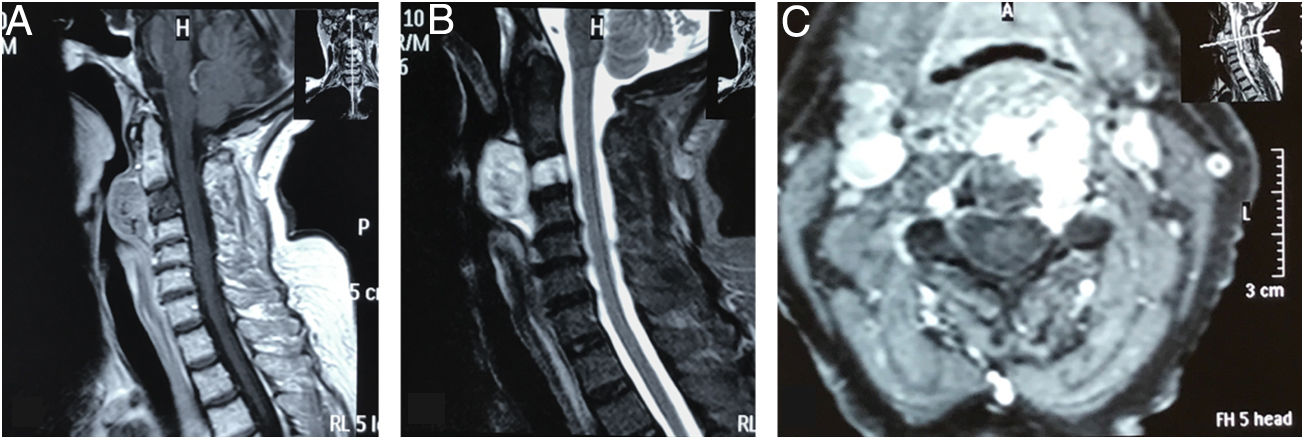
. Corte sagital ponderado en T1 que muestra lesión hipointensa en el cuerpo de C3, con extensión a la base de C2, masa tumoral prevertebral retrofaríngea desde C2 hasta el espacio C3-C4. Lesión con bordes hiperintensos de pequeño tamaño a nivel de la base de la odontoides. B) Corte sagital ponderado en T2 con masa prevertebral con realce, hiperintensa. Lesión del cuerpo de C3 hiperintensa. No evidencia de afectación discal. C) Corte axial de imagen con contraste a nivel de C3, que muestra compromiso tumoral hipercaptante que se proyecta al pedículo izquierdo y se insinúa en el conducto raquídeo.")
Resonancia magnética de columna cervical. A). Corte sagital ponderado en T1 que muestra lesión hipointensa en el cuerpo de C3, con extensión a la base de C2, masa tumoral prevertebral retrofaríngea desde C2 hasta el espacio C3-C4. Lesión con bordes hiperintensos de pequeño tamaño a nivel de la base de la odontoides. B) Corte sagital ponderado en T2 con masa prevertebral con realce, hiperintensa. Lesión del cuerpo de C3 hiperintensa. No evidencia de afectación discal. C) Corte axial de imagen con contraste a nivel de C3, que muestra compromiso tumoral hipercaptante que se proyecta al pedículo izquierdo y se insinúa en el conducto raquídeo.
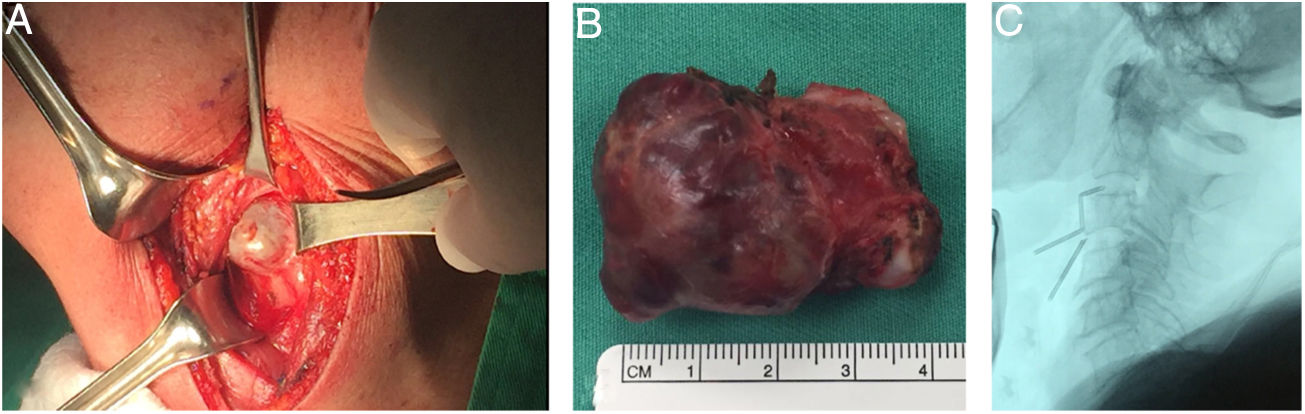
Fue llevada a resección quirúrgica mediante abordaje longitudinal anterolateral izquierdo, realizando hemicorpectomía ipsilateral de C3, disectomía C2-C3 y C3-C4, con resección tumoral en bloque de la masa prevertebral (fig. 3), estabilización espinal mediante cilindro intersomático y placa anterior de C2 a C4 (fig. 4). No se presentaron complicaciones postoperatorias. Las muestras de patología obtenidas intraoperatoriamente fueron categorizadas como Cordoma Convencional. En el postoperatorio, la paciente fue inmovilizada durante 45 días con collar cervical rígido tipo Philadelphia y posteriormente con collarín blando hasta completar los 2 meses POP.
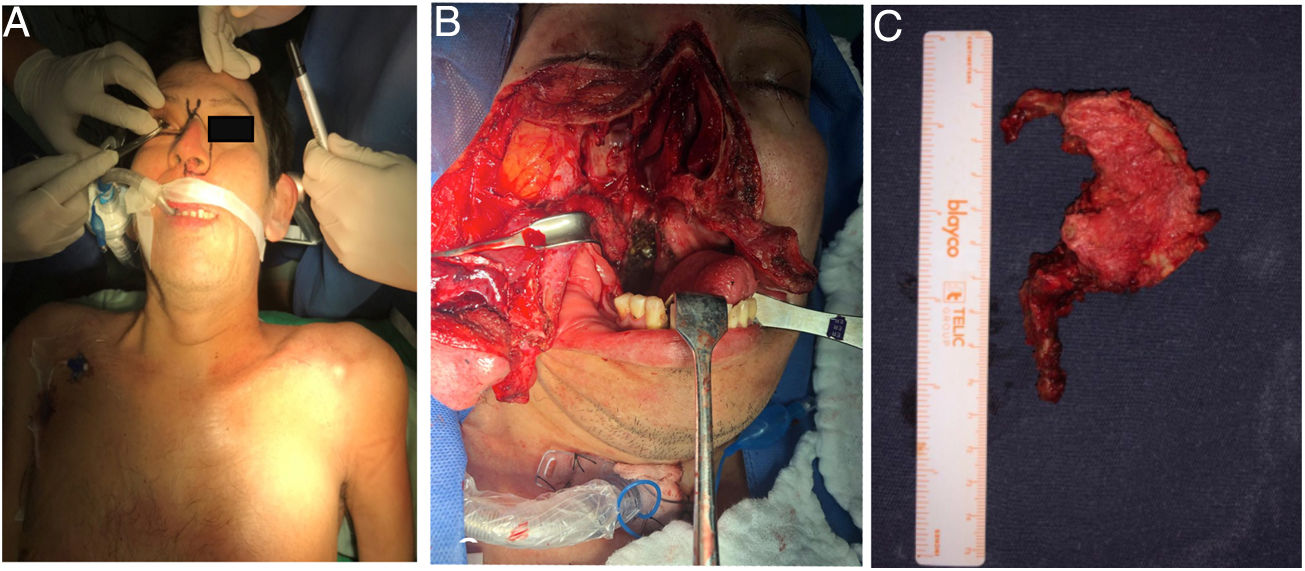
 Masa tumoral prevertebral vista mediante abordaje longitudinal anterolateral izquierdo. B) Espécimen tumoral de masa prevertebral. C) Identificación radiológica transoperatoria de nivel cervical.")
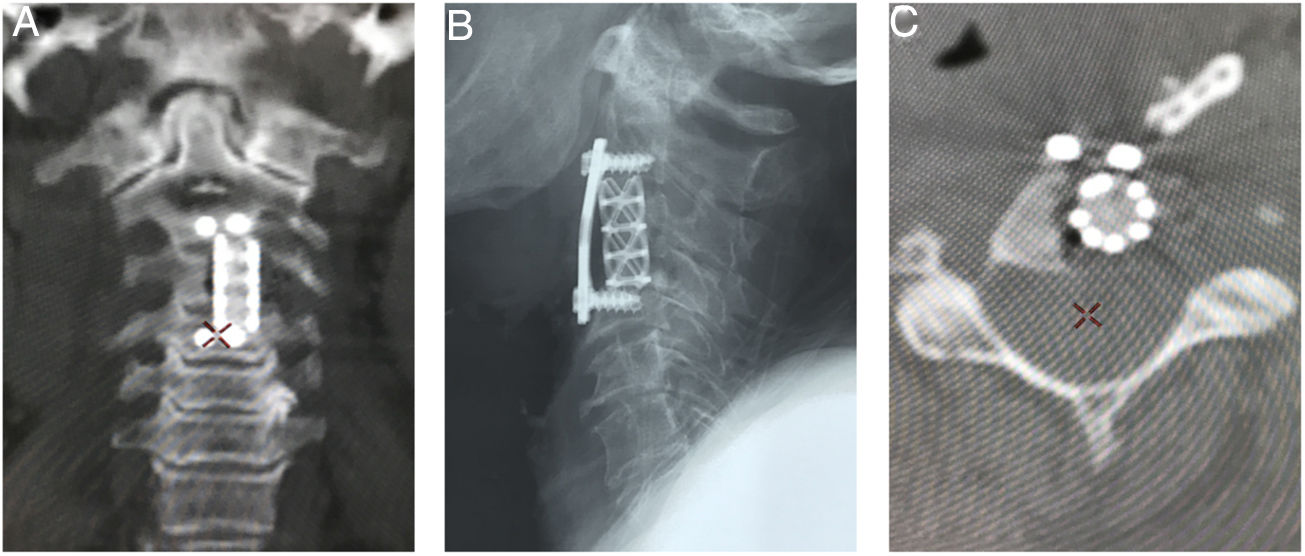
 Corte coronal de tomografía que muestra el soporte anterior intersomático mediante malla entre C2 y C4. B) Radiografía lateral que evidencia la estabilización cervical con el uso de placa anterior con fijación en C2 y C4. C) Corte axial de tomografía a nivel de C3 en donde se identifica la malla de titanio con aloinjerto (esponjoso liofilizado).")
Imágenes postoperatorias. A) Corte coronal de tomografía que muestra el soporte anterior intersomático mediante malla entre C2 y C4. B) Radiografía lateral que evidencia la estabilización cervical con el uso de placa anterior con fijación en C2 y C4. C) Corte axial de tomografía a nivel de C3 en donde se identifica la malla de titanio con aloinjerto (esponjoso liofilizado).
La paciente fue sometida a una cirugía de citoreducción y estabilización, seguida de radioterapia estereotáxica con gamma knife, iniciada a los 45 días de la intervención quirúrgica. Durante el seguimiento a 2 años POP, no fue evidenciada recidiva tumoral ni complicaciones relacionadas con los implantes, adecuado control del dolor con EVA 2/10 y ROM cervical sin limitación.
Caso 2Hombre de 41 años de edad, con antecedente traumático 7 meses previos a la consulta posterior a caída axial de 4 metros de altura, con alteración de la sensibilidad asociada tipo parestesias y sensación de debilidad. Acude a consulta externa por tetraplejía de 6 semanas de evolución, compromiso del estado general y nutricional.
A la exploración, función motora y sensitiva abolida a nivel C3, con hiperreflexia generalizada, Homman, clonus en miembros inferiores y Babinsky bilateral positivos. Sin control de esfínteres, con un calculó de índice de Karnofsky de 30/100.
El paciente aporta una radiografía de columna cervical de calidad insuficiente, realizada en un centro de atención primaria, que evidencia rarefacción ósea en unión craneocervical (fig. 5).

Se realiza TAC (fig. 6) y RMN simple de columna cervical (fig. 7) en las que se aprecia lesión tumoral a nivel de la unión craneocervical que se extienda caudalmente hasta C2.

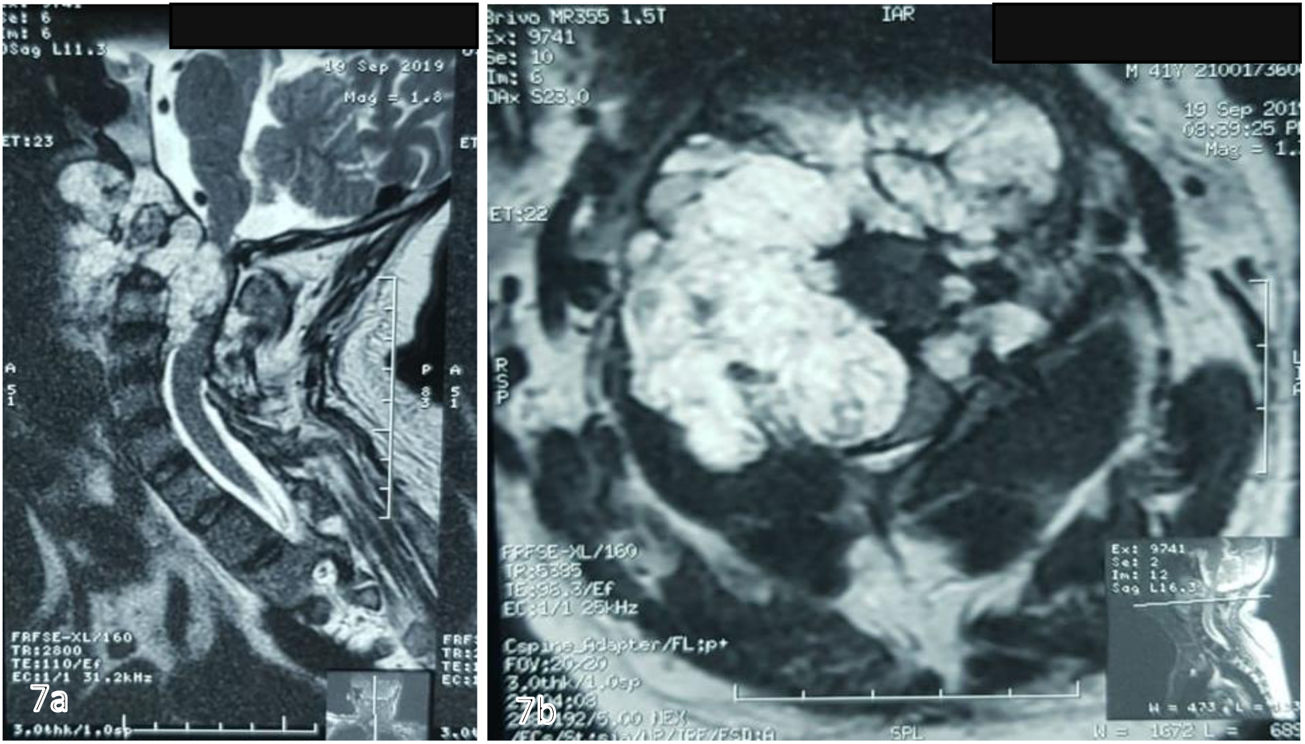
 Corte sagital ponderado en T2 que evidencia masa tumoral, lobulada hiperintensa que afecta clivus, C1, C2, retrofaringe e invade foramen magnum, generando compresión ventral severa del cordón medular. B) Corte axial ponderado en T2 en donde se aprecia masa lobulada voluminosa que ocupa retrofaringe y se proyecta posteriormente por el lado derecho, destruyendo masas laterales de C1 y C2, invade conducto raquídeo y musculatura paravertebral ipsilateral.")
RMN simple de columna cervical A) Corte sagital ponderado en T2 que evidencia masa tumoral, lobulada hiperintensa que afecta clivus, C1, C2, retrofaringe e invade foramen magnum, generando compresión ventral severa del cordón medular. B) Corte axial ponderado en T2 en donde se aprecia masa lobulada voluminosa que ocupa retrofaringe y se proyecta posteriormente por el lado derecho, destruyendo masas laterales de C1 y C2, invade conducto raquídeo y musculatura paravertebral ipsilateral.
No se evidenciaron alteraciones de laboratorio ni enfermedad metastásica en estudios de extensión. El paciente es sometido a tratamiento quirúrgico mediante doble abordaje quirúrgico en un solo tiempo.
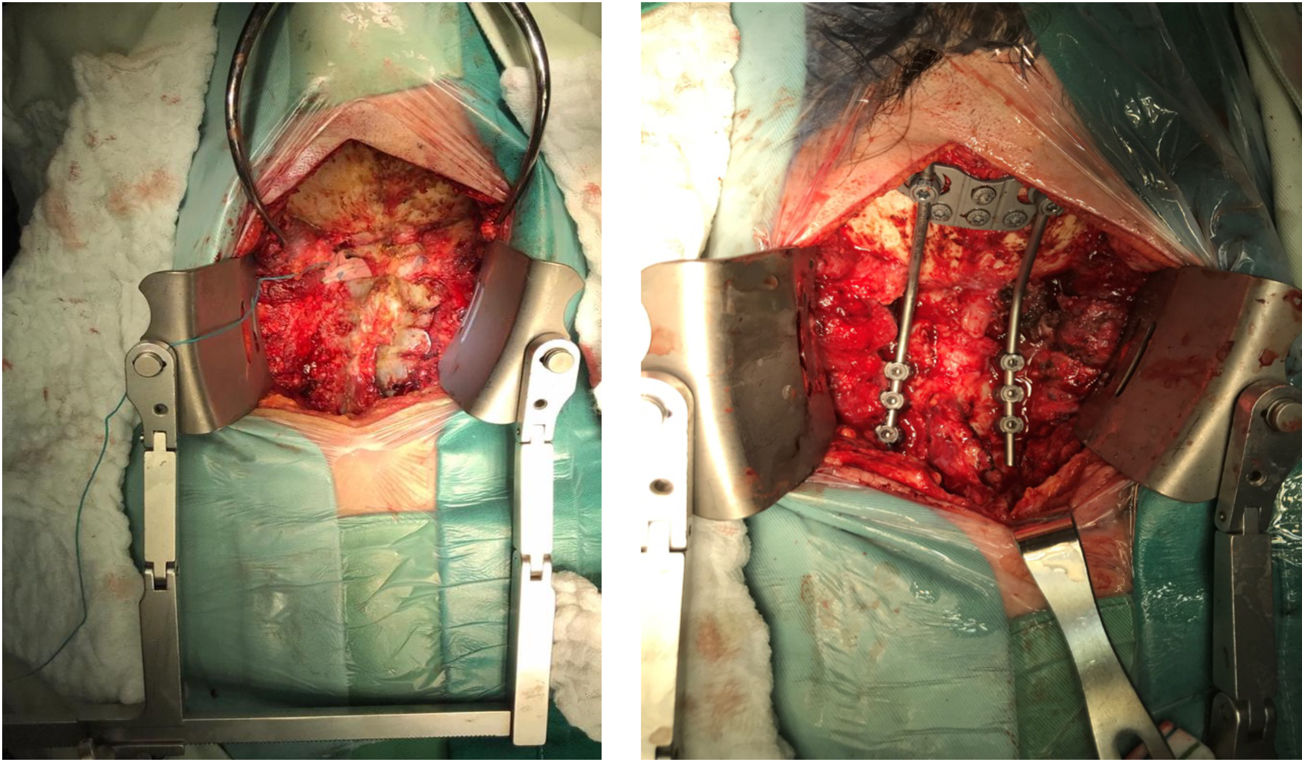
Por abordaje posterior se realiza descompresión con resección de arco posterior de C1 y lamina de C2 y resección de masa intraraquidea extradural. Estabilización de la región occipitocervical mediante instrumentación Occipito – C4, con tornillos de masa lateral (fig. 8).

Previa traqueostomía, por abordaje anterior transfacial bimaxilar orofaríngeo, se realiza cirugía de citoreducción con resección parcial de masa desde clivus hasta el cuerpo de C2. El abordaje facilitó la exposición predominantemente derecha, dada la localización tumoral y una descompresión satisfactoria del conducto raquídeo a nivel C1-C2 (fig. 9).
 Planificación de abordaje transfacial. B) Exposición de región craneocervical a través de región nasal y senos paranasales con incisión en pared posterior de rino y orofaringe. C) Espécimen patológico.")
En el postoperatorio, se inmoviliza con collarín rígido tipo Philadelphia, es trasladado a cuidado intensivo para soporte ventilatorio por 48 horas, donde se constata recuperación de sensibilidad y FM 1/5 en las 4 extremidades. Recibió nutrición enteral por SNG durante 5 semanas, antibioticoterapia biconjugada y cierre de traqueostomía al 7mo día del postoperatorio, previo egreso. Se continuó plan de rehabilitación al alta hospitalaria.
Las imágenes postoperatorias muestran la fijación occipitocervical, la descompresión del canal medular y al paciente en fase postoperatoria temprana (fig. 10).
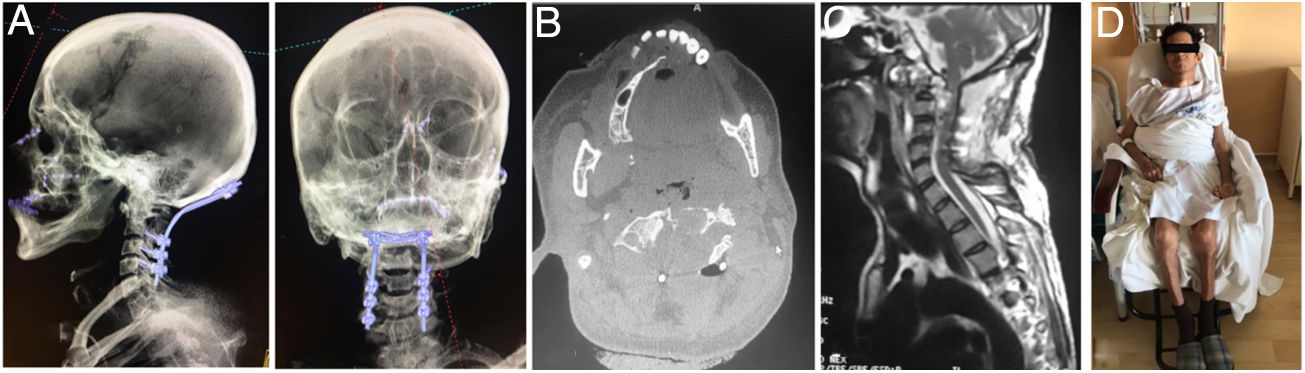
 Vista lateral y AP de reconstrucción 3D de tomografía en donde se aprecia la instrumentación occipitocervical hasta C4, con adecuada alineación sagital y posición facial al horizonte. B) Corte axial de TAC, que evidencia resección parcial del cuerpo de C2 y laminectomía para descompresión. C) Corte sagital de RMN ponderada en T2, mostrando descompresión importante subtotal del cordón medular y del espacio retrofarígeo. D) Paciente en postoperatorio con buena tolerancia a la sedestación.")
A) Vista lateral y AP de reconstrucción 3D de tomografía en donde se aprecia la instrumentación occipitocervical hasta C4, con adecuada alineación sagital y posición facial al horizonte. B) Corte axial de TAC, que evidencia resección parcial del cuerpo de C2 y laminectomía para descompresión. C) Corte sagital de RMN ponderada en T2, mostrando descompresión importante subtotal del cordón medular y del espacio retrofarígeo. D) Paciente en postoperatorio con buena tolerancia a la sedestación.
El resultado histopatológico confirmó el diagnostico de cordoma convencional, iniciándose por oncología clínica tratamiento con Imatinib combinado con RT. A los 8 meses de seguimiento, no hay evidencia de recidiva tumoral local, instrumentación estable, EVA 3-4.
DiscusiónLa notocorda empieza su formación durante la tercera semana de la gestación, originada en las células de la línea primitiva del embrión. Constituye el eje central del esqueleto axial y tiene un rol fundamentalmente inductor en la formación del sistema nervioso central y periférico. En su recorrido axial, discurre entre los cuerpos vertebrales desde los últimos segmentos coxígeos, hasta su extremo cefálico distal en el margen dural del cuerpo del esfenoides, caudal y posterior a la silla turca. Sufre un proceso de degeneración temprana durante el desarrollo embrionario, existiendo vestigios normales de la misma en los núcleos pulposos de los discos intervertebrales, siendo el factor Brachyury, un factor codificado por el Gen T, el principal partícipe en el desarrollo, mantenimiento y regresión de la misma5.
En sus extremos cefálico y caudal, la notocorda no conforma una estructura cilíndrica uniforme; presenta una morfología más compleja favoreciendo que los procesos de regresión durante la vida fetal no se realicen de forma homogénea a lo largo de todo su trayecto, permaneciendo restos celulares notocordales en estas localizaciones, base de la patogénesis del cordoma.
Caracterizado microscópicamente por Vyrchow en 1857 como un tumor vacuolado 6 y descrito por primera vez bajo su nombre actual por Ribbert en la década de 1890, quien fue el primero en hipotetizar el origen notocordal7, el Cordoma constituye un tumor primario de la columna vertebral raro, de carácter maligno de bajo grado8, que más frecuentemente se presenta entre la quinta y sexta décadas de la vida, con una media de 58.5 años, siendo solo el 20% de ellos en menores de 40 años9. Representa entre el 2 y el 4% de los tumores primarios del hueso.
Constituye un tumor limitado al esqueleto axial, aunque formas extraesqueléticas han sido anecdóticamente publicadas 10 y puede comprometer la base del cráneo, la columna móvil o el sacro. Algunas series sugieren que hasta el 90% de los mismos surgen en los segmentos distales de la columna. Se reporta el 32% de ubicación intracraneal, 29.2% en el sacro y el restante 32.8% en el resto de la columna11. The National Cancer Institute 12 mostró una frecuencia similar en estas tres localizaciones, luego de evaluar 400 casos en su serie de 2001.
Presentan una incidencia fuertemente relacionada con la edad, con 0.08/100,000 habitantes con una media de 60 años y de 1.4/10.000.000 en niños13. Tiene una relación de 2:1 hombre-mujer en las presentaciones espinales, a diferencia de la similitud en las cifras en los cordomas de la base del cráneo.
Muestra diferencias raciales, siendo excepcional en descendientes de negros africanos adultos y en las formas pediátricas es más común en hispanos y menos frecuente en blancos, comparado con la forma adulta14. Tiene un crecimiento lento, con una propagación tisular local constante que típicamente resulta en una progresión inexorable de la enfermedad. Son capaces, aunque de manera muy rara, de producir metástasis a distancia en pulmón, hueso o tejidos blandos15, estando hasta en el 5% de los pacientes al momento del diagnóstico, con un impacto en la sobrevida, calculada en una media de 24 meses16.
Basados en la historia natural de la enfermedad, la sobrevida general a 5 años es de 72% en EEUU y 61% en Europa. La sobrevida relativa específica por edad, es mejor en la población infantil con 69% en EEUU y 90% en Europa, en adultos jóvenes 78% y 74% respectivamente y en mayores de 65 años, 57% y 48%. La posibilidad de presentación de metástasis a lo largo del desarrollo de la enfermedad alcanza el 30%, sobre todo en las etapas tardías de la misma17.
Chambers et al (2014) evaluó la sobrevida a 10 años en pacientes con cordoma de diferentes orígenes, resultando trascendente la localización tumoral, observado mejores resultados en los cordomas craneales comparados con los otros: 59% vs 46%18.
Su apariencia macroscópica externa es suave, lobulada, con una coloración gris blanquecina19, con superficie mixoide y mucinosa, que parece bien definido entre el hueso y el tejido blando. Microscópicamente, se compone de nidos y cordones de células, con uno de dos patrones citológicos distintos. En el primero, la célula característica y patognomónica denominada physaliphorous (burbuja en griego), contiene un núcleo redondo con abundante citoplasma vacuolado. El núcleo está colocado excéntricamente con múltiples vacuolas o incluso una sola vacuola grande es más común en preparaciones histológicas de rutina. En el segundo, es un patrón constituido por células poligonales epitelioides con citoplasma eosinofílico. La atipia celular es escasa en los casos primarios y la actividad mitótica es inusual, y su presencia marca un comportamiento agresivo. La presencia de necrosis tumoral se reporta en cerca de un tercio de los casos siendo este un factor de mal pronóstico, así como la presencia de positividad del marcador Ki-67. El estroma intercelular característico está compuesto de un material mixoide y mucopolisacárido azul grisáceo20.
Su inmunohistoquímica permite diferenciarlo de los condrosarcomas y los adenocarcinomas mucinosos, los principales diagnósticos diferenciales. Es inmunoreactivo a la proteína S-100 y a los marcadores epiteliales como el EMA (Antígeno de membrana epitelial), las citokeratinas, el CEA (Antígeno carcinoembrionario), la alfa - antiquimitripsina y lisozima21.
En la última revisión sobre tumores de tejidos blandos y hueso en 2013 de la World Health Organization (WHO) y clásicamente, se distinguieron tres tipos de cordoma22. Hoch et al (2006), adiciona un subtipo denominado cordoma pobremente diferenciado23.
- 1.
Clásico o convencional: Caracterizado por un patrón celular con arquitectura lobulada y abundante matriz mixoide. Se considera el más frecuente y se presenta predominantemente en adultos. Representa hasta el 97% de todos los casos.
- 2.
Condroide: Similar al convencional, pero con una matriz que recuerda los tumores de origen de cartílago hialino. Presentan grandes rasgos celulares de desdiferenciación, con mayor capacidad para originar metástasis y presentar una transformación a una lesión de alto grado, típicamente a un histiocitoma fibroso, fibrosarcoma y menos frecuentemente, a osteosacoma o rabdomiosarcoma.
- 3.
Desdiferenciado: Con hallazgos sarcomatosos y bifásicos de tumor que muestran combinaciones de un cordoma clásico con sarcoma de alto grado. Es el tipo más agresivo. Ocurre en pacientes adultos y población pediátrica. En el componente de alto grado de este tipo de cordoma, el brachyury y la queratina está perdida al menos en parte24.
- 4.
Pobremente diferenciado: Diagnosticado principalmente en población pediátrica y muchas veces muestra un comportamiento de alto grado con una clínica agresiva. La pérdida del marcador INI1 constituye la característica esencial de esta variante25.
Dentro de la genética del cordoma, se documentan la deficiencia del gen homólogo de la tensina, fosfatasa, vías de señalización derivadas del factor de crecimiento plaquetario 26 y la sobreexpresión del factor T (brachyury), siendo este último el considerado más específico, por ser un factor requerido para el desarrollo de la notocorda durante la embriogénesis. Sin embargo, marcadores genéticos definitivos no han sido aún identificados, aunque se reportan algunos casos de asociación familiar27.
Estudios que soportan la evidencia más convincente de la hipótesis del origen notocordal, muestran el descubrimiento de una duplicación genética en la transcripción del factor Brachyury en cordomas familiares. Estudios de hibridación genómica mostraron duplicaciones únicas en la región 6q27 en estos casos, regiones que solo contienen el gen Brachyury, encontrándose sobreexpresado de manera única en los cordomas en comparación con otros tumores óseos y cartilaginosos28.
Debido al crecimiento lento tumoral, en las etapas iniciales de la enfermedad, se presenta como una entidad silente, presentando síntomas en etapas tardías. Las manifestaciones clínicas y el patrón de compromiso varían ampliamente según la localización. Como se mencionó anteriormente, su lento crecimiento permite grandes volúmenes tumorales antes del inicio de las manifestaciones, sobre todo cuando se presentan en las regiones distales29.
Los tumores con localizaciones en la columna móvil surgen habitualmente en la unión de los dos tercios anteriores con el posterior del cuerpo vertebral y comparados con las demás localizaciones, se presentan con un menor tamaño, en pacientes más jóvenes y con un inicio más temprano de la sintomatología.
Los cordomas de la base del cráneo y el clivus típicamente se presentan con cefalea o disfunción de los nervios craneales, más frecuentemente del VI (Abducens) y más infrecuentemente, los del clivus pueden manifestar rinorrea debido a fístulas de líquido cefalorraquídeo30.
En las localizaciones cervicales, el dolor no específico del cuello con los arcos de movilidad sobre todo con la flexión y las rotaciones es el síntoma más frecuente, pudiendo irradiarse a los hombros y las extremidades. Ocasionalmente se produce disfagia debido al efecto de masa anterior. Pueden invadir cranealmente causando disfunción de los nervios craneales bajos, así como compresión del cordón espinal o de las raíces emergentes, causando mielopatía o radiculopatía respectivamente31.
En las regiones torácica y lumbar el dolor local no específico es la queja principal y puede ser la causa de fracturas patológicas, con la consecuente radiculopatía o mielopatía.
Las lesiones sacras tienden a comprometer la región sacrocoxígea, sobre todo S4 y S5, crecen lentamente contenidas en la fascia presacra 32 y causan compresión gradual de vísceras abdominales con presencia de dolor, compresión rectal, constipación, retención urinaria, disfunción sexual, y en lesiones de gran tamaño es posible la palpación de una masa en la región glútea33.
Estudios muestran que el déficit neurológico es más frecuentemente visto en cordomas de la columna móvil que en los sacrocoxígeos34.
Radiológicamente, los cordomas son lesiones destructivas de la línea media. Su patrón radiográfico típicamente es permeativo y totalmente lítico, con una delgada zona reactiva osteoesclerótica presente ocasionalmente, con una erosión lenta tumoral con expansión a los tejidos blandos.
En el TAC, el componente mixoide asociado con baja atenuación es común, permitiendo la valoración y cuantificación de la destrucción ósea asociada al cordoma. Aparecen como masas hipodensas comparadas con el hueso sano, pudiendo mostrar calcificaciones distróficas irregulares. Las lesiones cervicales pueden tener componentes epidurales y paravertebrales prominentes asociados con alargamiento foraminal, simulando tumoraciones de la vaina nerviosa (Shwanoma). Las lesiones sacras frecuentemente se asocian a una gran masa presacra de tejido blando con calcificaciones tumorales amorfas. En las lesiones de la columna móvil, estas calcificaciones son menos frecuentes, pero sí es común el colapso vertebral. El compromiso del espacio discal y de múltiples niveles vertebrales es frecuente a diferencia de los osteosarcomas y condrosarcomas35, haciendo necesario descartar cuadros de infección. Esta forma de presentación ocurre hasta en el 50% de los pacientes36.
La RMN el es mejor método para su estudio y es fundamental para valorar la extensión tumoral y el grado de afectación de estructuras adyacentes incluyendo su extensión en el espacio epidural. En ella, aparecen como tumoraciones de baja intensidad en las señales ponderadas en T1, pudiendo mostrar imágenes focales de hiperintensidad, las cuales representan hemorragia intratumoral. La imagen ponderada en T1 con contraste con gadolinio demuestra una captación heterogénea del tumor con una apariencia de panal de abejas. En las imágenes ponderadas en T2, se observan como lesiones hiperintensas, debido a su rico contenido en mucina y matriz condroide. Las imágenes de supresión grasa ayudan con la delimitación de los bordes tumorales37.
El diagnóstico definitivo se obtiene mediante el estudio anatomopatológico. La biopsia de la lesión debe ser realizada bajo guía tomográfica, evitando la realización de biopsias abiertas siempre que sea posible, para evitar la contaminación de los márgenes tumorales. Sin embargo, en la unión craneocervical, debido a la presencia de estructuras nobles (vasculonerviosas) la técnica percutánea es altamente demandante. El consecuente cuidado en la resección del trayecto de biopsia durante el posterior acto quirúrgico, reviste gran importancia para evitar la posterior recidiva tumoral.
El curso insidioso de la enfermedad y la invasión tumoral a estructuras óseas y neurológicas críticas, hace que el manejo de estos pacientes sea desafiante y requiera su derivación a centros especializados que cuenten con oncólogos, cirujanos ortopedistas y neurocirujanos, patólogos, cirujanos reconstructivos y radioterapeutas, con experticia en el manejo.
La base del tratamiento consiste en una resección quirúrgica completa con márgenes negativos, sin embargo, no es muchas veces factible dada la proximidad a estructuras nobles38. Clásicamente, se considera que la resección incompleta, localización en la base del cráneo, edad avanzada, tamaño tumoral grande y necrosis intratumoral, son factores asociados con el pobre pronóstico39.
Recientemente, Zhou J et al (2017) describe los resultados de una serie retrospectiva de 682 pacientes con cordoma derivados de la experiencia institucional y la revisión de 108 estudios de la literatura, concluyendo que la localización espinal sobre todo sacra, edades jóvenes, patología desdiferenciada y antecedentes de QT constituyen predictores negativos para la sobrevida libre de tumor, mientras que a mayor edad, presentación con disfunción intestinal y/o vesical, patología desdiferenciada, recurrencia, progresión y presencia de metástasis determinan la peor sobrevida global40.
De manera global, se reconocen 4 modalidades de manejo para el cordoma:
- 1.
Quirúrgico aislado con resección total con márgenes amplios.
- 2.
Resección total con márgenes amplios o marginales + radioterapia (RT).
- 3.
Resección subtotal con márgenes intralesionales + RT.
- 4.
RT aislada.
El control macroscópico tumoral y la calidad de los bordes de resección son los factores más importantes en el control local de la enfermedad 41 y en el pronóstico de sobrevida42, por ello, la radioterapia efectiva cumple un papel trascendental en lograr el control de la enfermedad microscópica asociada al tratamiento quirúrgico43.
Bergh et al (2000), describió que la progresión del cordoma local se asocia significativamente con un mayor riesgo de metástasis y muerte relacionada con el tumor, por lo tanto, un mejor control local puede resultar en un mejor control sistémico y supervivencia, considerando que la RT adyuvante primaria temprana después de la cirugía tiene tasas más altas de control de la enfermedad que la hecha en el paciente derivado para el tratamiento de rescate de la enfermedad recurrente44. Hug et al (1995) demuestra la concordancia con los reportes de Bergh en el estudio de cohorte comparativa de pacientes con RT adyuvante temprana y aquellos con terapia de salvamento, describiendo un control local a 5 años del 88% vs 9% respectivamente, con mayores recurrencias en los tumores sacros (84.6%) vs no sacros (15.4%)45.
Radioterapia de intensidad modulada basada en fotones (IMRT): A pesar de la utilidad de la RT en el control del cordoma, la tolerancia de órganos y tejidos alrededor de la columna espinal, es un limitante en la dosis total de RT que puede ser usada. Los reportes antiguos, con uso de dosis que no exceden los 60Gy de RT convencional luego de la cirugía radical, reportan un control local pobre de los cordomas espinales46, con cifras de recurrencia de 50% a 100% y una sobrevida libre de tumor a 5 años de menos del 25%, considerando estos resultados como consecuencia de la relativa resistencia a la RT por parte del cordoma y a la imposibilidad para dar altas dosis sin afectar a los tejidos sanos con las terapias antiguas47.
Esta técnica permite modular el haz de fotones para que se adapte a las características tridimensionales del tumor, minimizando la dosis sobre los tejidos circundantes, concentrando la exposición sobre los tejidos tumorales, permitiendo así el aumento de dosis recibida.
Zabel et al (2010) en una serie de 34 pacientes con cordoma sacro tratados con IMRT, con una dosis media de 54Gy en 1.8Gy encontraron un control local significativamente mayor en los pacientes que recibieron dosis> 60Gy en comparación con las dosis totales <60Gy con efectos secundarios moderados de la radioterapia y los pacientes que recibieron> 60Gy mostraron una supervivencia general significativamente mejor (85% frente a 43%, P <0.01)48.
La IMRT puede ser usada mediante una fracción única de alta dosis o de manera hipofraccionada (<5 fracciones). Las dosis altas en única fracción causan un daño irreparable del DNA, aspecto importante debido a la relativa radioresistencia del cordoma mediante la habilidad de reparación del mismo y las altas dosis hipofraccionadas, actúan mediante el incremento en la apoptosis endotelial49.
Terapia con haz de protones: Como se ha mencionado, el objetivo de la radioterapia es lograr grandes dosis tumorales, con la menor cantidad de radiación recibida por el tejido sano, muchas veces extremadamente difícil, por la cercanía del cordoma con estructuras críticas. Esta preocupación aumenta ante la presencia de lesiones satélites que también deben ser incluidas en el volumen tumoral, incrementando la cantidad de radiación recibida. La RT con Protones tiene como objetivo abordar este desafío mediante la administración de una pequeña dosis al tejido normal sin afectar la dosis específica sobre el objetivo tumoral50.
Radioterapia con partículas de iones pesados: Comparada con la RT de protones, los iones de carbono tienen la ventaja biológica al incrementar la efectividad relativa debido a su habilidad para entregar una energía media mayor por unidad de longitud de su trayectoria, conocida como transferencia de energía lineal, evitando lesiones en órganos críticos inducidas por la radiación y demostrando tasas de control local muy cercanas a las obtenidas mediante IMRT.
Nishida et al (2011), mediante el estudio retrospectivo de 17 pacientes con cordoma sacro primario, tratados mediante cirugía aislada (10 pacientes) con media de seguimiento de 5 años y mediante RT aislada con carbono con una media de 70.4Gy (7 pacientes) con seguimiento de 4 años, mostraron un control local de la enfermedad de 62.5% para el grupo de cirugía y del 100% en el de RT, sin evidencia de toxicidad neurológica y manteniendo la función anorectal y urinaria postratamiento51.
La presencia de material a base de titanio en el interior del volumen objetivo puede afectar potencialmente la precisión de la RT al afectar la capacidad para delinear con precisión el tumor y reducir la cobertura homogénea52. A pesar de ello, estudios como los de Rotondo et al (2015) e Indelicato et al (2016), muestran tasas comparables de control local con RT pre y postoperatoria a los 5 años, con 82% y 88% respectivamente. 53–54.
En este momento, la radioterapia adyuvante con iones de carbono, haz de protones y con fracción única de fotones, tienen similares tasas de control local y sobrevida general, sin embargo, se requieren estudios que comparen los perfiles de toxicidad, dando información sobre la seguridad y eficacia de las diferentes modalidades55.
La resección quirúrgica de los cordomas espinales resulta la alternativa standard para el control macroscópico de la enfermedad, sin embargo, tiene implicaciones significativas en la morbilidad y la función del paciente. La resección en bloque para lograr bordes amplios se considera el método más adecuado para ofrecer los mejores resultados de control tumoral56. Este precepto es modificable para los cordomas localizados en el clivus, en los cuales, el énfasis está en la preservación de la función neurológica, llevando a cabo una cirugía de citoreducción segura con la posterior RT.
De acuerdo a la clasificación de Enneking, existen 4 tipos de resecciones tumorales57:
- 1.
Intralesional: La resección ocurre a través de la masa tumoral.
- 2.
Marginal: La resección se extiende hacia o a través de la zona reactiva que rodea el tumor, pero no lo viola.
- 3.
Amplia: El margen de la resección se realiza a través de tejidos sanos.
- 4.
Radical: Se crea un margen radical cuando el compartimento óseo o miofacial que contiene el tumor es resecado por completo.
En la columna móvil, la resección ampliada o marginal se logra solo en un 21% debido al compromiso epidural58, mientras que en los cordomas sacros la cifra asciende a un 35 - 75%, con el riesgo de déficit neurológico secundario a lesiones de las raíces nerviosas de S1-S2 y la posterior disfunción rectal o vesical59.
Para el planeamiento quirúrgico, los abordajes anteriores son los más directos para resecar las lesiones ventrales de la unión craneovertebral (UCV), al mismo tiempo permiten una buena descompresión del conducto raquídeo y colocar el injerto óseo. Existen varios tipos de abordajes que los comentaremos someramente a continuación:
- 1.
Abordaje transfaríngeo transoral: Permite la visualización de la línea media desde el clivus hasta el disco C2-C3, pero no está indicado si hay que realizar grandes liberaciones del saco dural o resecciones en el área de la arteria vertebral.
- 2.
Abordaje tipo labiomandibuloglosotomía media: Facilita una exposición más amplia, tanto a caudal como en sentido transversal.
- 3.
Abordaje transoral con maxilotomías: Es el que más amplia visulaización en el sentido cefalocaudal de la exposición permite, facilitando en sentido proximal llegar hasta el esfenoides.
- 4.
Abordaje retrofaríngeo cervical anterior alto: Permite una exposición más amplia de C1 y C2, sobretodo en sentido lateral. Hacia distal, con una pequeña modificación, puede exponer toda la columna cervical. Permite mayor facilidad para la colocación del injerto y la instrumentación. Se requiere siempre identificar y movilizar el nervio hipogloso.
Estos abordajes requieren en el postoperatorio inmediato uso de ventilación mecánica, apoyo nutricional por sonda nasogástrica o nutrición parenteral, cuidados de higiene oral con antisépticos locales y antiboticoterapia combinada. Se recomienda con frecuencia colocar un drenaje de líquido cefaloraquídeo a nivel lumbar inmediatamente antes del procedimiento quirúrgico en caso de requerirse una liberación amplia del saco dural. Ocasionalmente puede estar indicada la traqueostomía, sobre todo si existe un déficit neurológico previo que genere en el paciente dificultad respiratoria.
El clásico abordaje anterolateral del cuello, pre esternocleideomastoideo no accede la UCV, sin embargo, es ideal para los tumores de C3 a distal. Con una buena disección proximal se puede exponer el cuerpo de C2 con el fin de realizar la osteosíntesis.
En la región posterior, el abordaje longitudinal medial cráneo cervical permite descomprimir la UCV en el caso de que la compresión tumoral sea posterior. Es también muy útil para estabilizar este segmento con barras, tornillos y cerclajes, adicionándose injerto óseo. Debe tenerse especial cuidado durante la disección para no lesionar las arterias vertebrales en el intervalo occípito C1. Este abordaje frecuentemente constituye un complemento al abordaje anterior. Según la estabilidad mecánica lograda puede usarse en el postoperatorio un cuello rígido tipo Philadelphia o incluso un halo chaqueta.
Al día de hoy, no se ha establecido un tratamiento definitivo fuera de la cirugía. La quimioterapia no ha mostrado ser efectiva, de ahí que el enfoque haya cambiado en la actualidad hacia la investigación de la terapia molecular dirigida como modalidad alternativa60.
La aberración en la señalización molecular de la expresión del receptor de la tirosina kinasa (TKR) está asociada con la tumorogénesis. Muchos receptores, alteraciones genéticas, anormalidades cromosómicas y vías moleculares han sido recientemente identificadas para el mejor entendimiento de la patogénesis, y por ende del manejo de los cordomas. Estos receptores de TKR y vías de señalización molecular incluyen el factor de crecimiento epidérmico, el factor de crecimiento derivado de las plaquetas alfa y beta, AKT fosfoinositide – 3 – kinasa, activador mitógeno de la proteína kinasa entre otros61. Derivado de estos conocimientos, existen múltiples reportes retrospectivos de casos, series y estudios clínicos en fase 2, que soportan la utilidad y efectividad de inhibidores de los receptores, sin embargo, los resultados del uso de estos medicamentos resultan controversiales en la literatura.
Estos agentes no constituyen la primera opción de manejo para la entidad y solo están recomendados para cuadros avanzados o recurrentes que no responden al manejo quirúrgico o la RT, siendo sus resultados difícilmente evaluables en el cordoma. Los resultados clásicamente son evaluados siguiendo los criterios CHOI, que se basan en los cambios en el tamaño tumoral y en la densidad luego de la administración intravenosa de contraste para la toma de TAC o RMN62.
La terapia molecular dirigida en cordomas incluye:
- •
Imatinib y dasatinib contra los receptores del factor de crecimiento derivado de las plaquetas y el receptor del factor celular63,64.
- •
Erlotinib, lapatinib, gefitinib y cetuximab dirigidos contra los receptores del factor de crecimiento epidérmico y contra el erbB-2 (receptor tipo 2 del factor humano de crecimiento epidermal65.
- •
Sorafenib, pazopanib, y sunitinib que se dirigen contra factores angiogénicos como el receptor del factor de crecimiento endotelial vascular (VEGFR)66.
- •
Temsirolimus y sirolimus que se dirigen a la Fosfoinosotide 3 – kinasa67.
El imatinib fue el primer agente evaluado y es actualmente el más frecuentemente usado. La mayoría de pacientes con receptores para el factor de crecimiento derivado de las plaquetas se benefician de se uso para evitar la progresión de la enfermedad rápida. Dentro de los efectos adversos derivados de su uso se encuentran edema, anemia crónica, fatiga y hemorragia intraventricular subaguda68.
Hindi et Al (2015) en un estudio retrospectivo de 48 pacientes adultos con cordoma progresivo, localmente avanzado o metastásico que recibieron imatinib oral con una media de 600mg/día, por un periodo de 7 meses en promedio, evalúan la respuesta terapéutica demostrando enfermedad estable en el 74% de los casos (considerada como la mejor respuesta al tratamiento) y 26% con enfermedad progresiva, con el 20% de los pacientes tratados, libres de progresión de la enfermedad por más de 18 meses; considerando en tratamiento tumoral desafiante y demandante, con la necesidad de opciones terapéuticas diferentes para pacientes con enfermedad avanzada69.
Stacchiotti el al (2018) mediante un estudio de fase 2 con uso de Imatinib y everolimus en 43 pacientes con progresión avanzada del cordoma, evaluaron la tasa de respuesta general de acuerdo a los criterios CHOI, sobrevida libre de progresión y sobrevida general. Demuestran respuesta general de 55.8% de enfermedad estable, 20.9% con respuesta parcial y 16.3% con enfermedad progresiva, un tiempo libre de progresión de 9 meses en el 58.8% y de 12 meses en el 48.1%, encontrando toxicidad en más de la mitad de los pacientes, requiriendo retiro del tratamiento de manera definitiva en 30.2% y temporal en 60.5%, concluyendo que la respuesta médica farmacológica es limitada en cordomas avanzados70.
Como se evidencia en los dos estudios anteriores, existen cifras de inefectividad con su uso. En estos casos, los fármacos que actúan contra el factor de crecimiento epidérmico constituyen la segunda línea de manejo, dentro de estos el Erlotinib, el cual ha mostrado buenos efectos clínicos en cordomas refractarios al Imatinib71. Se considera que las terapias combinadas posibilitan la heterodimerización aumentando la sensibilidad farmacológica del cordoma hasta un 54%, siendo el caso, por ejemplo, del uso de la combinación entre gefitinib y cetuximab, reduciendo además los efectos secundarios de su uso de manera aislada72.
Otras anormalidades genéticas consisten en la pérdida de SMARC B1/INII y aberraciones en el microRNA y pérdidas o ganancias cromosomales73. En particular, la pérdida de 1p36ha sido asociada con pobre pronóstico. En este camino, Palbociclib está siendo estudiado como un inhibidor de CKD4/6 en el tratamiento del cordoma.
En conclusión, y ante el gran arsenal con el que actualmente cuentan lo oncólogos para el manejo farmacológico del cordoma bajo las condiciones antes mencionadas, la selección de las terapias moleculares dirigidas, deben estar basadas en el screening genético y la inmunohistoquímica. La monoterapia es la primera línea de recomendación y la combinación farmacológica, la opción ante cordomas resistentes.
La vacunación dirigida al factor Brachyury se encuentra bajo investigación, dado el conocimiento de su impacto sobre la patogénesis y la asociación familiar con casos de cordomas. GI-6301 es una vacuna recombinante derivada de levadura, codificante de brachyury, capaz de activar las células T humanas, promoviendo una respuesta inmune. Este agente induce la maduración de las células dendríticas del huésped, con la consecuente activación de células T CD4+ y CD8+ que se dirigen contra las células que expresan Brachyury, otorgando una acción antitumoral74.
Ha sido estudiada contra varios tipos de tumores sólidos incluyendo el cordoma y actualmente se encuentra durante la fase II de estudio, en pacientes con cordoma localmente avanzado e irresecable. Los resultados de la fase I han demostrado que es segura y bien tolerada. Los resultados de la fase II determinarán la eficacia clínica adicional75.
El cordoma constituye un tumor primario del hueso infrecuente, con predilección esquelética axial y con comportamiento agresivo por su capacidad de crecimiento local e invasión a tejidos adyacentes. Inicialmente indolente, muestra sintomatología en etapas avanzadas provocando según su localización, síntomas secundarios a compresión en estructuras vecinas que involucran el sistema nervioso central y periférico.
Requiere alta sospecha diagnostica para lograr un tratamiento temprano, constituyendo un gran desafío terapéutico médico y quirúrgico en búsqueda de una resección tumoral total con márgenes amplios. En aquellos casos en los que este objetivo no se logra, las modalidades de radioterapia recientes revisten utilidad trascendental para la reducción del riesgo de recidiva local, permitiendo grandes dosis dirigidas a la masa tumoral, con escaso impacto sobre tejidos sanos contiguos.
En la actualidad, se encuentra en desarrollo continuo el tratamiento médico encaminado a las alteraciones genéticas detectadas e incluyen fármacos dirigidos a la señalización de la tumorogénesis y la innovación de una vacuna activadora de células inmunes contra el factor Brachyury, dando una luz en el tratamiento adyuvante de casos difíciles por su localización, invasión sobre estructuras nobles y recidivas.
En nuestro medio, la dificultad para un diagnóstico precoz y para el acceso a RT, asociado a grandes tamaños tumorales infiltrantes, empobrece el pronóstico y tiene un alto impacto sobre la calidad de vida del paciente.
Fuentes de financiaciónRecursos propios de los autores.
Conflicto de interésLos autores no declaran algún conflicto de interés.
