




Se describe el caso de un paciente varón de 52años que consulta al servicio de reumatología por presentar adenopatías indoloras en las regiones cervical, axilar, supraclaviculares y en la nuca, así como fiebre, aumento de tamaño de parótidas y submaxilares. Se realizan estudios complementarios que arrojan como resultado anemia normocítica-normocrómica, trombocitopenia y eosinofilia, alteración de la función renal con hipoalbuminemia y hematuria, FAN 1/5.120, Sm+, ACL+ y biopsia de los tejidos comprometidos, por lo que se arriba al diagnóstico de enfermedad de Rosai-Dorfman y enfermedad relacionada con IgG4. Se discuten diagnósticos diferenciales de adenopatías cervicales, axilares e inguinales, fiebre, compromiso renal y hematológico.
The case is presented on a 52-year-old male patient, who was seen in the Rheumatology department. He had painless lymph nodes in the cervical, axillary, supraclavicular, and neck region. He also had a fever, and parotid and submaxillary gland enlargement. Complementary studies were performed, showing normocytic-normochromic anaemia, thrombocytopenia and eosinophilia, impaired renal function with hypoalbuminaemia and haematuria, ANA 1/5120, Sm+, ACL+. Biopsies were also performed on the compromised tissues, reaching the diagnosis of Rosai-Dorfman disease and IgG4-related disease. Differential diagnoses of cervical, axillary and inguinal lymph nodes, with fever, renal and haematological compromise are discussed.
La enfermedad de Rosai-Dorfman (ERD) es una rara entidad que se caracteriza por fiebre, adenopatías, neutrofilia, eritrosedimentación elevada e hipergammaglobulinemia policlonal. Los hallazgos anatomopatológicos se caracterizan por una infiltración histiocitaria y otras células inflamatorias, junto a la emperipolesis. Inmunohistoquímicamente muestra histiocitos con positividad para S100 y CD68; asimismo, es característico que los histiocitos sean negativos para CD1a. La enfermedad extranodal, comunicada en el 33-44% de los casos, se localiza con mayor frecuencia en la cabeza y en el cuello, mientras que la afectación renal se ha descrito en el 4% de los casos, asociada a peor pronóstico y una mortalidad del 40%.
La enfermedad relacionada con IgG4 (ER-IgG4) es una nueva entidad clínica, caracterizada por una concentración elevada de IgG4 en suero y tumefacción o infiltración de tejido por células plasmáticas positivas para IgG4, acompañada de fibrosis estoriforme. Puede manifestarse de manera similar a la enfermedad de Mikulicz, a la pancreatitis autoinmune, a la hipofisitis, a la tiroiditis de Riedel, a la neumonitis intersticial, a la nefritis intersticial, a la prostatitis, a la linfadenopatía, a la fibrosis retroperitoneal, al aneurisma aórtico inflamatorio y al seudotumor inflamatorio.
En la literatura se encuentran pocas comunicaciones de casos sobre la asociación de estas dos entidades, las cuales comparten características clínicas e histológicas. Se presenta el caso de un hombre de 52años con adenopatías cervicales múltiples, indoloras, con compromiso renal y de glándulas salivales, que constituyó un desafío diagnóstico. Se considera oportuno tener en cuenta estas enfermedades poco conocidas a la hora de hacer un diagnóstico correcto.
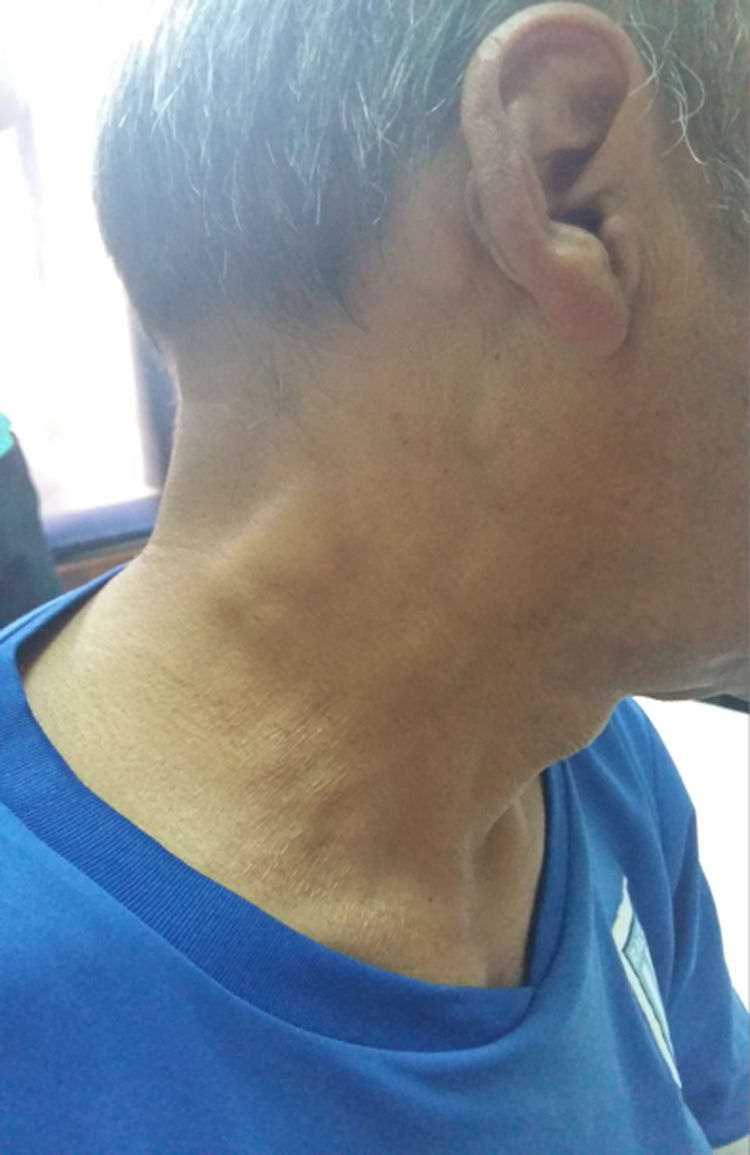
Presentación del casoPaciente masculino de 52años con antecedente de diabetes mellitus tipo 2 que consultó por presentar fiebre, adenopatías bilaterales indoloras en región cervical, nuca, axila y regiones supraclaviculares (fig. 1). También se halló aumento de tamaño de glándulas salivales: parótidas y submaxilares (fig. 2). No se apreciaron cambios visuales, síntomas sicca, rash malar, Raynaud, ni úlceras nasales u orales, pérdida de cabello, disfagia, disnea o tos. No había tampoco antecedentes familiares de trastornos neurológicos ni enfermedades tiroideas. El paciente negaba hábitos tóxicos, uso de drogas ilegales o alergia medicamentosa. Al examen físico se constató fiebre, adenopatías cervicales, axilares e inguinales, bilaterales, de 2-3cm de diámetro. Al examen cardiovascular y pulmonar no se hallaron alteraciones. El abdomen se encontró normal.


En el laboratorio el paciente exhibió anemia normocítica normocrómica, trombocitopenia y eosinofilia. Asimismo, eritrosedimentación y PCR aumentadas; C3 y C4 disminuidos; hipergammaglobulinemia policlonal; proteinuria 1.518mg (30-140mg/24h); creatinina en sangre 4,15mg/dl (rango normal=0,50-0,90mg/dl); urea 1,18g/l (rango normal=0,10-0,40g/l); hipoalbuminemia 1,5g/dl (rango normal=3,50-4,80g/dl) y hematuria; factor antinuclear (FAN hep2) 1/5.220 (histona) y anticuerpos anticardiolipinas (ACL) positivos; anticoagulante lúpico y beta 2 glicoproteína 1 negativos; IgG e IgE sérica elevadas; factor reumatoideo, anti-DNA ds, anti-Ro; La, RNP y ANCA P y C ELISA para PR 3 y mieloperoxidasa negativos; anticuerpos antitiroideos y hormonas tiroideas normales; IgG 4 e IgG séricas 1.376mg/dl (700-1.600mg/dl) y 948mg/l (110-1.570mg/l), respectivamente. Los cultivos de sangre y orina fueron negativos. Las evaluaciones para sífilis, virus de inmunodeficiencia humana, tuberculosis, toxoplasmosis, micoplasma, citomegalovirus, virus Epstein-Barr, hepatitis B y C fueron negativas.
Se realizó una biopsia renal cuyo resultado informó infiltración intersticial renal por elementos linfohistiocitarios, glomérulo con leve proliferación de células mesangiales, atrofia tubular 20% y nefritis intersticial. El paciente presentó fibrosis intersticial (no estoriforme) e infiltrados linfoplasmocitarios (predominio de linfocitos) sin eosinófilos. No se observó flebitis obliterante. Inmunomarcación: 15 células IgG4+ por alto campo, relación IgG4/IgG del 25%. Se realizó también biopsia de un ganglio cervical que presentó folículos linfoides con centros germinales. Las áreas interfoliculares se observaron expandidas, a expensas de numerosos plasmocitos y focos de emperipolesis. Inmunomarcación: CD68 positivo, S100 positivo, CD1a negativo, IgG4 100 células por alto campo y un rango IgG4/IgG: 45%.
En la biopsia de médula ósea se observó hipercelularidad con relación mielolipomatosa de 80/20, aumento de plasmocitos del 50%, CD138+ y focos de emperipolesis. La biopsia de glándula salival menor presentó infiltración linfoplasmocitaria, fibrosis leve e intersticial (no estoriforme), sin flebitis obliterante ni eosinófilos. Acúmulos linfocitarios periductales. Inmunomarcación, IgG4: 12 células por alto campo y un rango de IgG4/IgG del 25%. No cumple criterios diagnósticos de lupus eritematoso sistémico (LES), según los criterios ACR 1997, a pesar de presentar dichos anticuerpos.
Material y métodoSe llevó a cabo una búsqueda bibliográfica en libros de reumatología (Robert G. Lahita, George Tsokos, Jill Buyon, Takao Koike; Systemic Lupus Erythematosus. Fifth Edition. USA. Elsevier, 2011) y una búsqueda sistemática de bibliografía en bases de datos (MEDLINE, PubMed, LILACS) usando términos en español e inglés como «adenopatía», «enfermedad relacionada con IgG4», «histiocitosis» y «lupus eritematoso sistémico», sin fechas límites para los resultados.
Diagnóstico diferencialEl diagnóstico diferencial de un paciente con adenopatías cervicales, axilares e inguinales, fiebre y compromiso renal y hematológico incluye: LES, síndrome de Sjögren primario (SSs), ER-IgG4, enfermedad de Castleman, enfermedad de Kikuchi-Fujimoto, síndrome de PFAPA (fiebre periódica, estomatitis aftosa, faringitis y adenitis cervical) y ERD.
Lupus eritematoso sistémicoEn el LES, la linfadenopatía ocurre en un 50% de todos los pacientes durante la evolución de la enfermedad. Los grupos más comprometidos son los cervicales (43%), mesentéricos (21%), axilares (18%) e inguinales (17%). La linfadenopatía cervical es la primera manifestación clínica de la enfermedad en el 2% de los pacientes. Los mismos son típicamente no inflamatorios, blandos y discretos, varían de tamaño —de uno a varios centímetros— y se presentan en pacientes jóvenes. Histológicamente, los ganglios presentan focos paracorticales de necrosis e infiltración de linfocitos, histiocitos, células plasmáticas e inmunoblastos, con «cuerpos hematoxilínicos» que son clásicos de la linfadenopatía lúpica. La fiebre es un signo común de la actividad del LES, pero siempre debe considerarse la infección. Por ello, se excluyó este diagnóstico presuntivo, ya que la histología de nuestro paciente no mostraba dicha hiperplasia reactiva ni cuerpos hematoxilínicos, ni tampoco cumplía con los criterios clasificatorios para LES de ACR de 1982.
Otras enfermedades importantes se han asociado a linfadenopatías y LES, como la ERD, la enfermedad de Castleman, el linfoma y la enfermedad de Kikuchi. La linfadenitis necrotizante histiocítica del LES es patológicamente similar a la enfermedad de Kikuchi-Fujimoto. Estas dos entidades clínicas también pueden coexistir, o bien la enfermedad de Kikuchi puede preceder al diagnóstico de LES. La enfermedad de Castleman o hiperplasia angiofolicular del nódulo linfático es una enfermedad rara, de etiología desconocida, que puede presentarse como linfadenopatía local o multicéntrica. Esta linfadenopatía multicéntrica y otras características clínicas pueden imitar al LES1.
Enfermedad de Kikuchi-FujimotoLa enfermedad de Kikuchi-Fujimoto, también conocida como linfadenitis histiocítica necrotizante, es una enfermedad autolimitada de la mujer joven, de causa desconocida, la cual puede simular al LES. Los signos y los síntomas incluyen adenopatía cervical, fiebre, pérdida de peso y un pródromo de infección del tracto respiratorio superior. El diagnóstico se basa en los hallazgos histológicos de la biopsia. Las adenopatías cervicales son el principal sitio de la enfermedad y se ven en un 70-98% de los pacientes. Sin embargo, se ha descripto el compromiso de otras áreas nodales, incluyendo adenopatías periféricas (principalmente axilar) torácica, abdominal y pelviana. La fiebre es un síntoma primario en el 30-50% de los casos.
Aun cuando ciertas características clínicas se presentan comúnmente en esta entidad (linfadenopatía cervical, leucopenia, eritrosedimentación elevada y edad de menos de 30años), no hay nada específico que la sugiera2. Histológicamente, se caracteriza por la expansión paracortical de los ganglios linfáticos con áreas de necrosis irregulares y bien circunscritas que muestran abundantes restos nucleares cariorrécticos y ausencia de neutrófilos y eosinófilos. Se han reconocido 3 patrones histológicos en evolución: proliferativo, necrotizante y xantomatoso. Por inmunohistoquímica, los histiocitos son positivos para mieloperoxidasa. Hay un marcado predominio de células T en las lesiones (con células mayoritariamente positivas para CD8) y muy pocas células B1.
El diagnóstico histológico diferencial de la enfermedad de Kikuchi incluye otras causas de linfadenitis necrotizantes, específicamente infecciones, enfermedades autoinmunes y neoplásicas. La biopsia es importante para excluir malignidad y confirmar el diagnóstico2. Si bien nuestro paciente es masculino y presentaba hallazgos clínicos y de laboratorio compatibles con enfermedad de Kikuchi-Fujimoto, al revisar el compromiso histológico y la inmunohistoquímica dichos hallazgos no resultaron ser característicos de esta enfermedad.
Síndrome de SjögrenOtro de los diagnósticos que es preciso tener en cuenta es el SSs, una exocrinopatía autoinmune, caracterizada por ojo seco, boca seca y agrandamiento parotídeo. La complicación más preocupante es el desarrollo de un desorden linfoproliferativo, más comúnmente linfoma no Hodgkin. La linfadenopatía es un signo frecuente de esta enfermedad. Los pacientes pueden presentar un agrandamiento único o en múltiples sitios, pero la linfadenopatía masiva, con o sin esplenomegalia, en SSs es un signo de alerta acerca del desarrollo de un desorden linfoproliferativo. Igualmente, la linfadenopatía asociada a desorden linfoproliferativo requiere la presencia de células linfoides neoplásicas en la biopsia de médula ósea o del ganglio linfático.
Drosos et al. describen un paciente varón con SSs que desarrolló ERD. El paciente había ingresado al hospital con boca seca, agrandamiento bilateral de parótidas, linfadenopatías generalizadas y esplenomegalia3. La biopsia de glándula salival menor mostró una sialoadenitis focal con score de un foco según criterios de Chisholm y Mason, siendo el primer caso publicado en la literatura con esta asociación. El paciente había desarrollado una infección por Brucella, luego desarrolló un SSs y al poco tiempo histiocitosis sinusal con linfadenopatía masiva (HSLM). Se especula que una infección crónica como brucelosis y un desorden inmune como SSs pueden llevar al desarrollo de HSLM. Tampoco se puede excluir una asociación de estas dos entidades4. Si bien los síntomas sicca no se encontraban en nuestro paciente, al realizar biopsia de la glándula no se halló tampoco patrón histológico característico de SSs.
Síndrome antifosfolipídicoLos anticuerpos antifosfolípidos, presentes en este paciente (por única vez), han sido reportados en pacientes con nefritis lúpica y en aquellos sin otras características de enfermedad sistémica (e.g., síndrome antifosfolipídico). A pesar de la presencia de títulos elevados de ACL en el paciente, este no presentó características clínicas de síndrome antifosfolípido y tuvo un solo dosaje sérico de ACL y anticoagulante lúpico. El paciente tuvo muchas manifestaciones clínicas de LES: linfadenopatías generalizadas, fiebre inexplicable y pérdida de peso. El diagnóstico se confirma por la presencia de anticuerpos en suero. Aunque el FAN puede verse en otras enfermedades autoinmunes, los anticuerpos anti-DNA nativo y anti-SM son relativamente específicos para el diagnóstico.
La causa de fallo renal agudo asociado a síndrome nefrítico es menos obvia. Aunque dicha causa suele ser aparente, este fallo inexplicable es una indicación de biopsia renal. La nefritis intersticial no puede excluirse, debido a la asociación de piuria con fallo renal agudo. El compromiso intersticial es común en la nefritis lúpica, y la fibrosis intersticial extensa tiene una estrecha relación con el fallo renal progresivo. Los medicamentos antiinflamatorios no esteroideos —recibidos por este paciente— pueden causar nefritis intersticial. También consideramos una nefritis intersticial alérgica por la cefalosporina que el paciente había recibido, necrosis tubular aguda sobre la base de hipoalbuminemia y síndrome antifosfolipídico1.
InfeccionesDentro del espectro de las enfermedades infecciosas deben descartarse: tuberculosis, yersiniosis, toxoplasmosis, enfermedad por arañazo del gato y linfogranuloma venéreo, entre otras. La infección localizada es la causa más frecuente de fiebre y linfadenopatía cervical. La forma más común de infección localizada es la faringitis, que puede ser viral, producida por micoplasma o piógena y debido a estreptococo, estafilococo aureus y estafilococo epidermidis. La tonsilitis debido a una variedad de organismos puede presentarse como linfadenopatía cervical. La linfadenitis cervical tuberculosa (escrófula) es históricamente la causa más común de tuberculosis extrapulmonar1. Si bien siempre se las tiene en cuenta como posibles causas, estas fueron excluidas debido a serologías y cultivos negativos.
Enfermedad de Rosai-DorfmanLa histiocitosis sinusal con linfadenopatía masiva (HSLM), que también se conoce como enfermedad de Rosai-Dorfman (ERD), es un trastorno proliferativo histiocítico raro, de etiología desconocida, inicialmente descrito por Rosai y Dorfman en 19695. La Organización Mundial de la Salud (OMS) y la Histiocyte Society la incluyen en el grupo II de la Clasificación de las Histiocitosis y Enfermedades de las Células Dendríticas, que engloba a las histiocitosis de fagocitos mononucleares distintos de las células de Langerhans, en contraposición con la histiocitosis de células de Langerhans (grupo I) y las histiocitosis malignas (grupo III)6.
La ERD es una histiocitosis sinusal con linfadenopatía masiva que afecta predominantemente a chicos. Presenta también infiltrados masivos, bilateral del tejido blando orbitario y linfadenopatía cervical extensa. Formas mínimas que afectan los párpados y la conjuntiva pueden ocurrir en adultos. Se caracteriza por linfadenopatía indolora con fiebre, leucocitosis, neutrofilia, eritrosedimentación elevada e hipergammaglobulinemia policlonal. La forma de presentación más frecuente de esta entidad consiste en adenopatías cervicales bilaterales masivas e indoloras7. Como se puede observar, las que presentaba nuestro paciente, acompañadas por los resultados de laboratorio, tenían compromiso renal intersticial y de glándulas salivales.
La hipótesis etiológica de la ERD incluye desórdenes en la regulación inmune, al igual que infecciones causadas por agentes como el virus del herpes, virus Epstein-Barr, citomegalovirus, Brucella y Klebsiella. Las anormalidades inmunológicas y su significación en la ERD han sido reportadas por Foucar et al. en 230 casos de HSLM. De estos casos, 23 fueron identificados con hallazgos clínicos y de laboratorio que sugieren un desorden inmune. Asimismo, se hallaron 9 pacientes con autoanticuerpos, 3 con glomerulonefritis, 2 con síndrome de Aldrich/Wiscot, 9 con manifestaciones musculoesqueléticas, 3 con infecciones inusuales y 6 con alteraciones musculares4.
SarcoidosisEs un trastorno multisistémico de etiología desconocida que afecta con mayor frecuencia a adultos de entre 20 y 40años. Los pacientes con sarcoidosis suelen presentar linfadenopatía hiliar bilateral, infiltración pulmonar y a menudo lesiones oculares y cutáneas. Se arriba a un diagnóstico de esta entidad cuando los hallazgos clínicos y radiográficos están respaldados por evidencia histológica de granulomas de células epitelioides no caseificantes encontrados en la biopsia de tejido. Un gran número de los pacientes (40-50%) con sarcoidosis crónica tienen compromiso extrapulmonar que puede afectar a casi cualquier tejido. Los granulomas sarcoides se infiltran comúnmente en los ganglios linfáticos y en las glándulas salivales.
Cuando se incluyen los ganglios hiliares, la incidencia de linfadenopatía en pacientes con sarcoidosis probablemente se aproxima al 90%, en tanto que la incidencia de linfadenopatía periférica es de alrededor del 27%. La linfadenopatía puede asociarse con enfermedades agudas y crónicas y generalmente es indolora. La linfadenopatía cervical es la manifestación más común de sarcoidosis en cabeza y cuello, pero la sarcoidosis solo representa el 1,7% de todas las linfadenopatías de cabeza y cuello6. La actividad de la enzima convertidora de angiotensina (ECA) en suero no parece aumentar el valor predictivo de las pruebas de función pulmonar y las radiografías torácicas, y no se recomienda su uso en la investigación para sospecha de sarcoidosis7. Nuestro paciente no tenía enfermedad pulmonar, ni erupciones cutáneas (e.g., eritema nudoso y lupus pernio), molestias musculoesqueléticas o disfunción neurológica (parálisis del nervio facial, dolor de cabeza y encefalopatía). De la misma manera, su ECA fue negativa, por lo que se alejó del diagnóstico.
Enfermedad relacionada con IgG4La ER-IgG4 es una nueva entidad clínica caracterizada por una concentración elevada de IgG4 en suero y tumefacción o infiltración de tejido por células plasmáticas positivas para IgG4. Puede manifestarse con una amplia variedad de entidades, a saber: enfermedad de Mikulicz, pancreatitis autoinmune, hipofisitis, tiroiditis de Riedel, neumonitis intersticial, nefritis intersticial, prostatitis, linfadenopatía, fibrosis retroperitoneal, aneurisma aórtico inflamatorio y seudotumor inflamatorio8.
La linfadenopatía concomitante, común en esta enfermedad, generalmente toma 2 formas. Primero, por medio de la participación de los ganglios linfáticos como enfermedad localizada adyacente a un órgano específico afectado por ER-IgG4. En segundo lugar, la linfadenopatía generalizada puede ser el único componente de las presentaciones clínicas en ER-IgG4.
Al parecer, los hallazgos histomorfológicos de la linfadenopatía relacionada con IgG4 mostraron diversidad histológica. Además, desde el punto de vista clínico, esta entidad solo ocasionalmente mostró linfadenopatía sistémica, hipergammaglobulinemia policlonal, en particular elevación de IgG e IgE, y positividad de varios autoanticuerpos. La enfermedad en mención se puede caracterizar en 5 subtipos histológicos: morfología similar a la enfermedad de Castleman (I), hiperplasia folicular reactiva (II), plasmacitosis e inmunoblastosis interfolicular (III), transformación progresiva del centro germinal (IV) y seudotumor inflamatorio (V)9.
Los pacientes con linfadenopatía sistémica relacionada con IgG4 eran significativamente mayores (68,8 vs. 43,3 años) y tenían concentraciones apreciablemente más bajas de proteína C reactiva (0,29 vs. 8,71mg/dl) e interleucina (IL) -6 (8,45 vs. 34,82pg/ml) que los pacientes con enfermedad de Castleman multicéntrica.
Uno de los diagnósticos diferenciales más importantes es la proliferación linfoplasmocítica atípica e inmunoblástica (linfadenopatía asociada a la enfermedad autoinmune). Koo et al. reportaron una lesión inusual en los ganglios linfáticos, «ALPIB», asociada con varias enfermedades autoinmunes, incluidas la artritis reumatoide y el LES. Histológicamente, se caracteriza por una infiltración linfoplasmocítica policlonal prominente con varios números de inmunoblastos. Sin embargo, no hay evidencia de enfermedad autoinmune definida en ninguna de las linfadenopatías relacionadas con IgG4. Los trastornos linfoproliferativos reactivos (LPD) atípicos o malignos que se presentan en pacientes inmunodeprimidos por metotrexato (MTX) para el tratamiento de la artritis reumatoide se han reportado desde principios de los años noventa. Un porcentaje de la linfadenopatía inducida por MTX parece ser similar a ALPIB10.
Cuando los linfomas angioinmunoblásticos de células T (AITL) contienen algunas células tumorales (células claras), con numerosas células plasmáticas e inmunoblastos B, pueden confundirse con las lesiones de tipo III. A diferencia de AITL, no hay células T CD10+ citológicamente atípicas y no se presenta proliferación dendrítica folicular extrafolicular en las lesiones de tipo III. Además, el AITL suele involucrar síntomas sistémicos.
Aunque algunos casos de linfadenopatía se designaron previamente como trastornos linfoproliferativos atípicos que simulan linfomas malignos, carecen de monoclonalidad del gen de la inmunoglobulina y se considera que no son neoplásicos. Los pacientes con linfadenopatía relacionada con IgG4 deben tener biopsias de ganglios linfáticos para excluir linfoma, sarcoidosis, enfermedad de Castleman multicéntrica o neoplasias diseminadas11.
Síndrome PFAPAEl síndrome de adenitis cervical, estomatitis aftosa, fiebre periódica, faringitis de comienzo en el adulto (PFAPA), junto con otras entidades como síndrome de Behcet, pericarditis idiopática recurrente, enfermedad de Still del adulto, artritis idiopática juvenil de comienzo sistémico, pertenece al grupo de enfermedades autoinflamatorias adquiridas (AID) sobre una base multifactorial o poligénica. El acrónimo PFAPA (fiebre periódica, estomatitis aftosa, faringitis y adenitis cervical) fue acuñado junto a los criterios diagnósticos de 1989, 2años después de la primera descripción de esta entidad realizada por Marshall en 1987. Esta entidad se caracteriza por fiebre alta (>39°C) asociada con al menos uno de los 3 signos clínicos cardinales: estomatitis aftosa, faringitis o adenitis cervical. Ha sido bien descripta en pacientes pediátricos, en general antes de los 5años, y es la causa más frecuente de fiebre periódica de origen desconocido en chicos.
En la literatura se han descripto cerca de 40 casos de comienzo en adultos. Las características clínicas de los pacientes adultos son similares a aquellas descriptas en los chicos, con excepción de que las artralgias y las mialgias parecen ser más frecuentes en los adultos que en los niños, mientras que las aftas y los escalofríos parecen ser menos comunes en los adultos. Los 3 signos cardinales (aftosis, faringitis y adenitis cervical) estuvieron presentes concurrentemente durante los ataques en el 47% de los adultos, en contraste con el 27% de los niños. Al menos 2 signos cardinales se vieron en el 52% de los pacientes adultos, en comparación con el 46% de los chicos. Asimismo, el número medio de episodios/año fue de 8,3±5,2 en los adultos y de 13,3±9,2 en la población de PFAPA. Se tomó dicho diagnóstico diferencial debido a que el paciente presentó adenopatías cervicales y fiebre, aunque no cumplía todos los criterios de inclusión.
Existen reportes de casos sobre la combinación del síndrome PFAPA con manifestaciones autoinmunes como glomerulonefritis proliferativa endocapilar, hepatitis autoinmune tipo 2, encefalitis aséptica aguda recurrente y parapsoriasis aguda. Estas asociaciones podrían ser ocasionales, hasta hoy no hay datos para establecer si la desregulación en PFAPA podría predisponer a estos pacientes a la autoinmunidad. No hay pruebas diagnósticas para este síndrome. Durante los ataques los pacientes pueden presentar moderada leucocitosis con predominio de neutrófilos. La eritrosedimentación y la proteína C reactiva están constantemente aumentadas durante los ataques febriles12. En este paciente, sin embargo, se descartó por la ausencia de aftas o faringitis y la presencia de leucocitosis con neutrofilia.
Deficiencia de adenosina deaminasa 2La deficiencia de adenosina deaminasa 2 (DADA 2) es una enfermedad autosómica recesiva descripta en 2014. Inicialmente reconocida como un síndrome que se manifiesta con fiebre, poliarteritis nudosa, livedo racemosa, accidente cerebrovascular de comienzo temprano e inmunodeficiencia mínima. La presentación clínica y la edad de comienzo varían ampliamente y las manifestaciones más severas son aplasia medular, aplasia pura de células rojas, neutropenia, enfermedad hepática y daño neurológico. Su presentación clínica es altamente variable. El comienzo de la enfermedad suele darse en la infancia, aunque se ha reportado en el adulto. El paciente de más edad tenía 59años y se presentó con ulceraciones en las piernas. La linfoproliferación es otra característica importante de esta entidad.
Los pacientes se presentan la mayoría de las veces con linfadenopatía generalizada (>10%) y esplenomegalia (>30%). Recientemente se han descripto casos con un fenotipo similar al síndrome linfoproliferativo autoinmune (ALPS) y otro paciente con una enfermedad similar a Castleman, asociada con fiebre, esplenomegalia, linfadenopatías generalizadas, anemia, trombocitosis, hipergammaglobulina policlonal e IL-613. En este paciente se descartó tal diagnóstico debido a la edad y a la falta de compromiso vascular y neurológico, aun cuando presentaba compromiso hematológico.
Enfermedad de KimuraLa enfermedad de Kimura es un trastorno inflamatorio de etiología desconocida que afecta principalmente a sujetos de origen asiático14. Desde el punto de vista clínico, suele manifestarse por la presencia de masas de tejido subcutáneo y subplastimal, localizadas por lo general en la zona de la cabeza y el cuello (76%), con participación de glándulas salivales y adenopatías regionales, como se pudo ver en nuestro caso. A nivel histológico, se caracteriza por la afectación de los nódulos linfáticos en forma de seudotumores fibroinflamatorios, con hiperplasia de folículos germinales, fibrosis intersticial, marcada infiltración eosinófila de la masa y de los tejidos adyacentes y, de manera frecuente, eosinofilia con aumento de IgE en sangre. También se destaca que comparte con esta entidad características histológicas halladas en la biopsia de ganglio cervical, pero no la característica hiperplasia de folículos ni tampoco la eosinofilia, como tampoco masas subcutáneas, por lo que se descartó la enfermedad en mención.
Diagnóstico clínico del discursorNuestro paciente presenta las características clínicas de una ERD, debido a que se encontraron linfadenopatías cervicales bilaterales y masivas, indoloras, acompañadas de fiebre, leucocitosis, gammapatía policlonal y velocidad de sedimentación globular acelerada, con afectación extranodal (compromiso de médula ósea y renal). Histológicamente, el paciente presentó una característica de la enfermedad, como lo es la emperipolesis y la inmunomarcación CD68+, S100+ y CD1a−. Además, la histología de los ganglios presentaba folículos linfoides con centros germinales, numerosos plasmocitos con células IgG4+, al igual que en riñón, médula ósea y glándula salival menor. Por lo tanto, habiendo sido descartadas otras causas de adenopatías cervicales, se asumió que el paciente presentaba una ERD y una patología autoinmune asociada (ER-IgG4).
Discusión anatomopatológicaEn la biopsia de ganglio linfático se hicieron hallazgos principalmente morfológicos inespecíficos, comúnmente asociados a linfadenopatías reactivas, como la hiperplasia folicular o la expansión del área interfolicular. Debido a la expansión del área interfolicular a expensas de plasmocitos, se emplearon técnicas de inmunohistoquímica para IgG e IgG4 y se constató un número de plasmocitos IgG4+ mayor de 100 células por campo de alto aumento, así como una relación IgG4/IgG del 45%. Ambos indicadores apoyan el diagnóstico de ER-IgG4. La ER-IgG4 en ganglio linfático se comporta como una linfadenopatía reactiva con distintos tipos de presentación según la arquitectura; en este caso podría haberse tratado del tipo II (símil a hiperplasia folicular reactiva) o del III (símil a expansión interfolicular)15. Sin embargo, la presencia de senos subcapsular y medulares que contienen histiocitos CD68+, S100+ y CD1a− con emperipolesis confirma el diagnóstico de ERD16.
Por otra parte, los hallazgos histológicos hechos tanto en la biopsia de glándula salival como en la biopsia renal son relativamente inespecíficos y no invalidan el diagnóstico de ERD. Si bien en ambos casos se observan más de 10 plasmocitos IgG4+, la relación IgG4/IgG es menor del 40%, por lo que se desestima el diagnóstico de ER-IgG4. Puesto que tanto la ERD como la ER-IgG4 pueden afectar múltiples órganos y estar asociadas a enfermedades autoinmunes, el diagnóstico puede ser dificultoso. Esto es especialmente cierto en aquellos casos de ERD con incremento de plasmocitos IgG4+, ya que alrededor de un 30% de los pacientes con ERD presentan más de 10 plasmocitos IgG4+ por campo de alto aumento y una relación IgG4/IgG mayor del 40%, por lo que ciertos hallazgos se solapan17.
La distinción entre ambas enfermedades es importante porque la ER-IgG4 tiene altas tasas de respuesta a corticoesteroides, mientras que en la ERD la respuesta es más bien baja en aquellos casos que no logran autolimitarse16. Algunos autores plantean la hipótesis de que los casos de ERD con respuesta a corticoesteroides podrían ser aquellos con un número aumentado de plasmocitos IgG4+, pero no existen estudios que lo demuestren17.
A diferencia de la ER-IgG4, la ERD no presenta fibrosis estoriforme ni flebitis obliterante, pero sí histiocitos S100+. Existen discrepancias entre las bases biológicas que diferencian a una enfermedad de la otra. Así, algunos autores señalan que se trata de enfermedades diferentes, ya que la cantidad de linfocitos T reguladores FOXP3+ es significativamente mayor en la ER-IgG4. Sin embargo, otros autores llegan a resultados contradictorios, puesto que observan el mismo fenómeno en casos de ERD extranodales, mas no en aquellos que afectan el ganglio linfático18.
Comentarios y resultado definitivoAunque la ERD puede presentarse a cualquier edad, es más común en el niño y en el adulto joven, con un leve predominio en el varón. Sin embargo, la edad a la que se presentaron los casos reportados con afectación renal (4%) fue claramente mayor, la mayoría en el rango entre la cuarta y la novena década de la vida. La etiología es desconocida. Se han descripto alteraciones del sistema inmune o agentes infecciosos, como pueden ser los agentes símiles del virus del herpes, del virus del Epstein-Barr, del citomegalovirus, de la Brucella o de la Klebsiella.
La ERD tiene un curso que se caracteriza por remisiones y exacerbaciones en la mayoría de los pacientes. Sin embargo, aquellos que presentan anormalidades inmunológicas antes o después de la manifestación de la enfermedad tienen un pronóstico menos favorable y una mayor mortalidad.
La histopatología de los tejidos comprometidos muestra emperipolesis, característica distintiva de esta entidad. Los histiocitos son CD1a negativos pero S100 positivos19. Algunos casos de ERD muestran muchas células plasmáticas IgG4 positivas, lo que produce confusión diagnóstica con la ER-IgG4. Con las características clásicas de linfadenopatías masivas y la evidencia de histiocitosis sinus, así como las características histopatológicas observadas en el nódulo linfático, en la médula ósea y en la lesión renal, se hace un diagnóstico de ERD. Aunque la ausencia de examen histológico impide asegurarlo, es muy probable que la esplenomegalia se deba a infiltración histiocitaria.
Si bien la ERD clásica se presenta con linfadenopatías cervicales masivas, bilaterales e indoloras, aproximadamente el 40% de los casos de ERD tienen compromiso extranodal o en otros tejidos, en algunos casos sin linfadenopatía asociada, la cual puede o no desarrollarse durante el curso posterior de la enfermedad. Los sitios extranodales más frecuentemente afectados son la piel y los tejidos blandos, el tracto respiratorio superior y el hueso, seguidos por el tracto genitourinario, el tracto respiratorio inferior, el ojo, el páncreas y la cavidad oral. En la ERD con afectación renal es preciso hacer el diagnóstico diferencial con el histiocitoma fibroso maligno y las proliferaciones histiocitarias de etiología infecciosa (la presencia de S100 es útil para diferenciarla de estas lesiones), leucemias y linfomas, especialmente cuando se acompañan de linfadenopatías (la ausencia de emperipolesis y el perfil inmunohistoquímico ayudan a un correcto diagnóstico).
El diagnóstico puede ser dificultoso y demorarse debido a una mala interpretación de los datos clínicos e histológicos, es realmente un desafío y puede ser equivocado e indicar sarcoidosis, artritis idiopática juvenil, espondiloartropatía o nódulos subcutáneos metastásicos. Otras patologías con las cuales se debe establecer el diagnóstico diferencial son las enfermedades por depósito, la tuberculosis, el carcinoma renal y las metástasis renales, sobre todo el melanoma20. El diagnóstico de la enfermedad en este caso se realizó por la clínica, la histología y el análisis inmunohistoquímico del nódulo linfático.
En general, la ERD tiene un buen pronóstico, aunque su evolución es impredecible y puede variar desde la remisión espontánea hasta la enfermedad persistente con episodios de remisión y exacerbación. Entre el 5 y el 11% de los pacientes fallece como consecuencia de la enfermedad, casi siempre son casos con alteraciones inmunológicas asociadas (anemia hemolítica autoinmune, glomerulonefritis, FR o FAN positivos). La afectación renal se ha asociado a un peor pronóstico, con una mortalidad asociada al 40%, aproximadamente. El compromiso extranodal, renal, de hígado y de tracto respiratorio inferior está asociado a peor pronóstico, al igual que el número de órganos extranodales afectados21. La manifestación extranodal más común o las aéreas más afectadas son cabeza y cuello en un 22% de los casos. La órbita o los párpados se ven afectados en un 10% de los casos.
En el laboratorio puede hallarse anemia y leucocitosis. El 90% de los pacientes presenta aumento de la velocidad de eritrosedimentación e hipergammaglobulinemia policlonal. Con menor frecuencia se detectan anticuerpos antinucleares. Aproximadamente el 13% de los pacientes tiene alteraciones autoinmunes concomitantes, como lupus eritematoso, artritis reumatoide y anemia hemolítica autoinmune22. Esta última, el hallazgo más frecuente, en algunos casos resulta fatal23.
En la literatura se encuentran 2 reportes que describen ERD en pacientes con LES. Se presenta el caso de un paciente con LES y síndrome anticuerpo antifosfolipídico que tuvo compromiso de glándula lagrimal, órbita, nodal y extranodal relacionado con la ERD. La asociación de la ERD con el LES es muy rara; solamente hay 3 casos informados en la literatura24. Juskevicius y Finley reportaron el caso de un hombre de 48años con LES y agrandamiento persistente de la glándula parótida derecha. La histología fue diagnóstica de ERD25.
Petschner et al. describen el caso de una mujer de 59años con LES que presentaba poliartralgias, mialgias, fotosensibilidad, poliserositis y adenopatías cervicales, axilares e inguinales. Esta paciente tenía FAN, complemento bajo y ACL IgM26. Como se ha comentado, la emperipolesis es característica de esta enfermedad, pero se puede encontrar en otras enfermedades, como 1) malignidades hematológicas: leucemia aguda, leucemia mieloide crónica, enfermedad de Hodgkin, linfoma no Hodgkin y síndromes mielodisplásicos y mieloproliferativos; o 2) malignidades no hematológicas: mieloma múltiple, neuroblastoma y rabdomiosarcoma27.
En fechas recientes se ha investigado la posible relación entre ERD y enfermedad esclerosante relacionada con IgG4, puesto que ambas entidades comparten rasgos histopatológicos como son el infiltrado linfoplasmocitario y la fibrosis estromal18. La patogénesis de la ERD no es clara. Existe evidencia de que puede asociarse con una respuesta autoinmune anormal. La mayoría de los casos de esta entidad mostró abundantes células plasmáticas y esclerosis. Varios reportes de caso y recientemente 2 series de un número limitado de casos han demostrado que las células plasmáticas se expresan en la ERD extranodal.
Zhang et al. observaron que una parte significativa de la ERD presenta características similares a la ER-IgG4, lo cual indica que podría existir superposición entre las dos entidades. En este estudio el número de células TREG fue comparable entre ERD nodal y los controles, mientras que la ERD extranodal mostró un número significativamente más alto de células TREG que los controles. Esto demostraría que un subgrupo de ERD tiene características de ER-IgG4, lo cual es indicativo de una superposición entre ciertos aspectos de estas dos entidades28.
En el caso del paciente reportado en este escrito se realizó inmunohistoquímica de los tejidos ganglionares, renales y salivales, en la que una alta proporción de células plasmáticas fue positiva para células IgG4. Chen y Lee, por su parte, observaron la presencia de fibrosis estromal con abundantes células plasmáticas, lo que sugiere ER-IgG4 como diagnóstico diferencial. En el compromiso extranodal se objetivan varios grados de fibrosis estromal29. El marcador de esta entidad es la proliferación intrasinusal de histiocitos S100, lo que demuestra emperipolesis y células inflamatorias, como en el caso del paciente reportado aquí.
La ERD comparte características histológicas con la ER-IgG4, a saber:
- •
Abundantes células inflamatorias.
- •
Varios grados de fibrosis estromal que en algunas ocasiones, como en los casos de compromiso extranodal, enturbian la distinción entre estas dos entidades.
La enfermedad de ERD es parte del diagnóstico diferencial de la ER-IgG4, dada su superposición histopatológica, pero se desconoce la verdadera relación entre estas. En la actualidad existe una amplia gama de estrategias terapéuticas disponibles. En el paciente asintomático puede indicarse observación y control o resección quirúrgica si hay riesgo de compresión de un órgano vital. De un 20 a un 50% de los pacientes con enfermedad cutánea/nodal pueden tener remisiones espontáneas, lo cual es aplicable en los casos de enfermedad cutánea/linfadenopatía no complicada y en aquellos con enfermedad asintomática en otros sitios.
Algunos casos en los que se ve amenazada la función de un órgano en forma grave o persistente pueden requerir tratamiento con cirugía, esteroides (la dosis y la duración no están definidos), quimioterapia (reservada para casos con recaída o refractarios, algunas veces se usa como terapia inicial cuando hay riesgo de vida o enfermedad diseminada) o radioterapia (de modesta eficacia en esta enfermedad, pero puede ser beneficiosa en enfermedad con compromiso de tejido blando refractario, compromiso orbital o enfermedad ósea con compromiso visual u obstrucción resistente de la vía aérea). Foucar et al. en 1990 describen 17/238 (7%) pacientes que murieron como resultado de complicaciones directas de la enfermedad, infecciones o amiloidosis. Por su parte, Pulsoni et al. en el 2002 describieron que 10/80 (12%) murieron como resultado de la enfermedad. Pacientes con enfermedad multifocal y extranodal, particularmente aquellos con enfermedad del tracto respiratorio inferior, hígado y riñón, tienen un mal pronóstico.
Los pacientes con ERD requieren exámenes clínicos y de laboratorio cada 3-6 meses durante los primeros 2años después de la remisión completa30.
Consideraciones éticasLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
