




El síndrome antifosfolípido se asocia frecuentemente con lupus eritematoso sistémico y otras enfermedades autoinmunes. Sin embargo, la coexistencia con vasculitis primaria ha sido poco reportada. Se presenta el caso de una paciente de 67años de edad con historia de aborto recurrente y tromboembolismo pulmonar crónico, quien es admitida para estudio de hemoptisis. A la evaluación inicial se diagnosticó una hemorragia alveolar masiva y glomerulonefritis. El resultado de los anticuerpos fue positivo para anticuerpos anticitoplasma de neutrófilos (ANCA) con patrón tipo perinuclear, anticuerpos anti-mieloperoxidasa y anticuerpos antifosfolípidos (anti β2 glicoproteína1 IgG y anticoagulante lúpico), configurándose el diagnóstico de vasculitis asociada a ANCA de tipo poliangitis microscópica en asociación con síndrome antifosfolípido. Dado el contexto clínico, se decidió iniciar metilprednisolona intravenosa en pulsos por 3días consecutivos, seguida de prednisona oral, y como terapia de mantenimiento se instauró rituximab y anticoagulación con warfarina. La evolución clínica de la paciente fue satisfactoria, alcanzando control de síntomas e importante mejoría de la función renal y pulmonar, con disminución del score BVAS.
Antiphospholipid syndrome is frequently associated with systemic lupus erythematosus and other autoimmune diseases. However, coexistence with primary vasculitis has been poorly reported. The case is presented of a 67-year-old patient with a history of recurrent abortion and chronic pulmonary thromboembolism who was admitted due to haemoptysis. At the initial evaluation, a massive alveolar haemorrhage and glomerulonephritis were diagnosed. The results of the antibodies were positive for ANCA with P-type pattern, anti-myeloperoxidase antibodies, and antiphospholipid antibodies (anti-β2 IgG glycoprotein1 and lupus anticoagulant). Diagnosis of ANCA positive vasculitis-type microscopic polyangiitis was made in association with antiphospholipid syndrome. Given the clinical context, it was decided to initiate intravenous methylprednisolone in pulses for 3 consecutive days, followed by oral prednisone, and as maintenance therapy, rituximab and anticoagulation with warfarin were instituted. The clinical evolution of the patient was satisfactory, with symptom control being achieved, as well as a significant improvement of renal and pulmonary function, with a decrease in the Birmingham vasculitis activity score (BVAS).
El síndrome antifosfolípido (SAF) es una condición definida por la coexistencia de trombosis venosa o arterial, o morbilidad gestacional en presencia de anticuerpos antifosfolípidos (anticoagulante lúpico, anticardiolipina o anti β2 glicoproteína1) persistentemente positivos por más de 12semanas1. El SAF frecuentemente se asocia con lupus eritematoso sistémico u otra enfermedad autoinmune subyacente, como anemia hemolítica, púrpura tombocitopénica idiopática, artritis reumatoide, síndrome de Sjögren, esclerosis sistémica, enfermedad mixta del tejido conectivo, polimiositis, entre otros2; sin embargo, la coexistencia con vasculitis sistémica primaria es infrecuente3.
La poliangitis microscópica (PAM) es una vasculitis necrosante, pauciinmune, que afecta predominantemente a vasos pequeños (p.ej., capilares, vénulas o arteriolas)4. Su inicio es más frecuente en mayores de 50años y afecta más a hombres que a mujeres; los dos órganos típicamente afectados y que a menudo definen el pronóstico son el riñón y el pulmón; sin embargo, puede afectar concomitantemente a otros órganos, como el sistema nervioso, piel, sistema musculoesquelético, corazón, ojos y tracto gastrointestinal5.
Una búsqueda realizada en Pubmed sobre la coexistencia entre PAM y SAF demostró un solo caso publicado en esta base de datos.
Se presenta el caso de una paciente con diagnóstico de vasculitis anticuerpos anticitoplasma de neutrófilos mieloperoxidasa (ANCA-MPO) positivo tipo PAM con compromiso pulmonar y renal, e historia de abortos recurrentes y tromboembolismo pulmonar crónico, configurándose el diagnóstico de SAF por la presencia de anticuerpos antifosfolípidos.
Caso clínicoUna mujer de 67 años es admitida para estudio debido a cuadro de 2meses de evolución de hemoptisis no masiva y fiebre subjetiva que aparece en el último mes, sin otros síntomas concomitantes. Al inicio del cuadro actual fue valorada en otra institución, donde realizaron una angiotomografía de tórax, la cual reveló trombos segmentarios recanalizados y áreas de vidrio esmerilado tanto en lóbulos superiores como inferiores; se realizó diagnóstico de neumonía y se decidió instaurar tratamiento antibiótico. Una semana después ingresa a nuestra institución. Su historial médico fue relevante por el antecedente de hospitalización 2años antes por un cuadro de hemorragia alveolar difusa confirmada por fibrobroncoscopia y una biopsia transbronquial que reveló signos de tromboembolismo pulmonar en la histopatología, sin evidencia de capilaritis; dado esto, se inició manejo con rivaroxabán (adicionalmente recibía suplemento con levotiroxina por hipotiroidismo primario). Ella no tomaba otros medicamentos ni tenía otros diagnósticos conocidos. Su historial obstétrico fue importante, dado que reportó 7 abortos embriogénicos, con más de 3 que ocurrieron de forma consecutiva antes de la semana10. En la revisión por sistemas se encontraron parestesias tipo adormecimiento en las plantas de ambos pies; otros síntomas fueron negativos.
Al ingreso se encontraba estable hemodinámicamente, con PA 104/65mmHg; FC 79/min; FR 20/min; SpO2 96% aire ambiente; peso 74kg. El resto del examen físico solo fue positivo por palidez en mucosas y crépitos finos en ambas bases pulmonares.
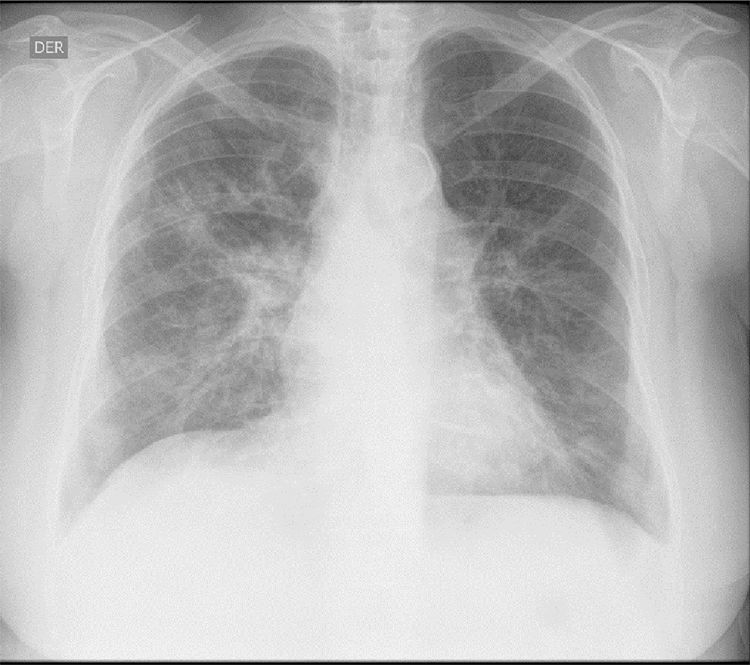
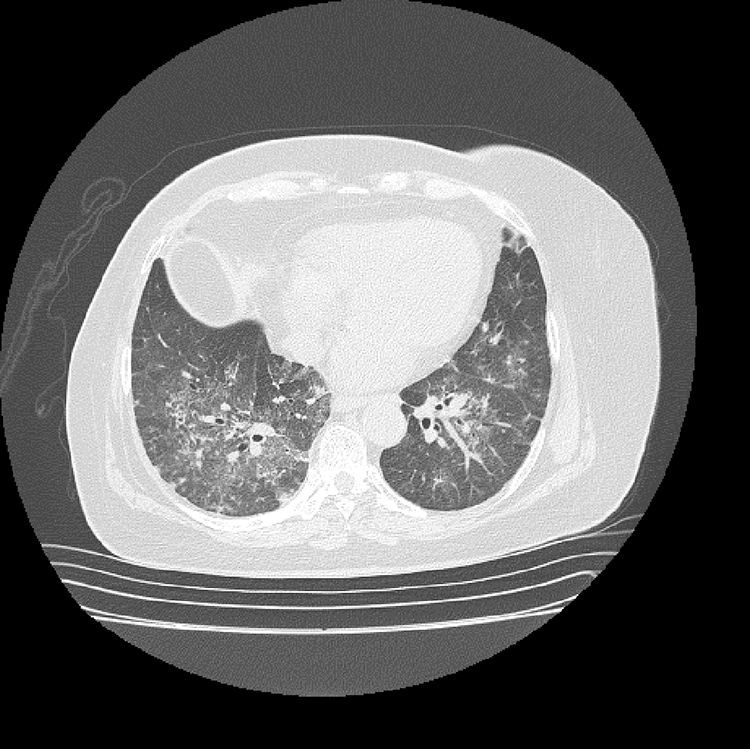
Los hallazgos de laboratorio iniciales revelaron una anemia moderada de volúmenes normales, elevación de la proteína C reactiva (PCR) y un sedimento urinario activo con proteinuria en rango no nefrótico. Los resultados de las ayudas diagnósticas realizadas se muestran en la tabla 1; la radiografía y la tomografía de tórax se muestran en las figuras 1 y 2, respectivamente.
Resultado de paraclínicos
| Examen | Ingreso | Día 4 | Día 7 | Día 18 |
|---|---|---|---|---|
| Hemograma | Leucocitos 6.460; Neutrófilos 4.500; Linfocitos 1.400; Hb 7,7; VCM 87,5; HCM 26,8; RDW 14,7; Hto 23%; Plaquetas 443.000 | |||
| Coagulación | TTP: 32,7; TP: 14,7; INR 1,1 | |||
| Creatinina | 1,32 | |||
| BUN | 24,5 | |||
| PCR | 39 | |||
| NA | 142 | |||
| K | 3,48 | |||
| Uroanálisis | Densidad 1.020; Proteínas 150mg/dl; Eritrocitos 250 (normal 0-5). Sin dismorfia eritrocitaria. Bacterias+++ | |||
| Baciloscopia en esputo | Negativa para BAAR | |||
| Proteínas en orina de 24h | 1.760 mg | |||
| Coombs directo | Negativo | |||
| C3 | 163 | |||
| C4 | 24,8 | |||
| Factor reumatoide | <8,0 | |||
| AG-HBS | Negativo | |||
| ANTI-HVC | Negativo | |||
| VDRL | No reactivo | |||
| Nivel IgE | 96,47 (alto) | |||
| Nivel de IgM | 69 (normal) | |||
| Nivel de IgG | 1.314 (normal) | |||
| Nivel de IgA | 319 (normal) | |||
| Anti-RO | <3,0 | |||
| Anti-LA | <3,0 | |||
| Anti-SM | 4,9 (negativo) | |||
| Anti-RNP | 3,1 (negativo) | |||
| Anti-ADN | Negativo | |||
| ANCA | Positivo 1/640 patrón perinuclear (ANCA-p) | |||
| Anticuerpos mieloperoxidasa (MPO) | 90,1 (positivo) | |||
| Anticuerpos proteinasa 3 (PR3) | 0,64 (negativo) | |||
| Anticuerpos membrana basal glomerular (GBM) | Menor de 1/20 | |||
| Anticuerpos antinucleares (ANA) | Negativo | |||
| β2-glicoproteína IgM | 1,4 (negativo) | |||
| β2-glicoproteína IgG | >100 (positivo) | |||
| Anti-cardiolipina IgM | 6,6 (negativo) | |||
| Anti-cardiolipina IgG | <3,0 (negativo) | |||
| Anticoagulante lúpico | Positivo |


A la paciente se le diagnosticó una hemorragia alveolar difusa y glomerulonefritis. Se solicitaron anticuerpos anticitoplasma de neutrófilos (ANCA) por IFI, los cuales resultaron positivos a título de 1/640 con un patrón perinuclear, el cual se confirmó con la prueba de Elisa, que fue positiva para anticuerpos mieloperoxidasa (MPO). Se realizó una biopsia renal con hallazgo de glomerulonefritis proliferativa extracapilar pauciinmune y la electromiografía demostró polineuropatía sensitiva de tipo axonal en los miembros inferiores, configurando el diagnóstico de vasculitis ANCA-MPO positivo tipo PAM (en ausencia de granulomas) con alta actividad de la enfermedad (BVAS 12/33 puntos). Los anticuerpos antinucleares (ANA) fueron negativos y de los anticuerpos antifosfolípidos resultó positivo los anti β2 glicoproteína1 IgG en título alto, además del anticoagulante lúpico, siendo negativos los demás. Basados en los antecedentes obstétricos, el hallazgo de tromboembolismo pulmonar por histología y angiotomografía de tórax, en presencia de anticuerpos anti β2 glicoproteína1 en título alto, se hizo el diagnóstico de SAF, teniendo en consideración la necesidad de repetir los anticuerpos antifosfolípidos a las 12 semanas, los cuales salieron positivos.
La paciente fue tratada con pulsos de metilprednisolona intravenosa durante 3días consecutivos, seguidos de prednisona oral a dosis de 1mg/kg/día. Como terapia de mantenimiento se instauró rituximab, presentando una marcada mejoría de la función renal y pulmonar, con disminución del score BVAS a 2/33. La anticoagulación durante el tiempo de hospitalización se realizó con heparinas de bajo peso molecular, y previo al egreso se inició warfarina con un objetivo de INR entre 2 y 3.
DiscusiónLa PAM es una vasculitis de pequeño vaso asociada a ANCA. Completan este grupo la granulomatosis con poliangitis (GPA) y la granulomatosis eosinofílica con poliangitis4.
La afectación en la PAM es multisistémica; la mayoría de los pacientes presentan compromiso renal; dos tercios tienen compromiso musculoesquelético; la mitad tienen lesiones en piel; la hemorragia alveolar ocurre en hasta en el 30% de pacientes, y en alrededor de un tercio de ellos se presenta compromiso gastrointestinal y neurológico5.
Describimos el caso de una paciente con vasculitis asociada a ANCA-MPO que reúne las características de PAM según el consenso de Chapel Hill actualizado en 2012, con compromiso renal dado por glomerulonefritis proliferativa extracapilar pauciinmune, demostrado en la histología; hemorragia alveolar difusa y neuropatía periférica, en ausencia de asma, eosinofilia e inflamación granulomatosa, lo cual la diferencia de otras vasculitis asociadas a ANCA4,5.
Tal como en esta paciente, los ANCA en la PAM están dirigidos predominantemente contra MPO, y se reconoce a partir de numerosos estudios su papel patogénico en esta condición, aunque los mecanismos por los cuales se desarrollan y se rompe la tolerancia permanecen desconocidos6. En una minoría de casos los ANCA pueden estar dirigidos contra proteinasa3 (PR3), aunque su rol en la patogenia es menos convincente7.
En la etiología de las vasculitis asociadas a ANCA se reconocen factores tanto genéticos como ambientales (p.ej., exposición a sílice, medicamentos como propiltiouracilo e infecciones, en particular por S.aureus)8, ninguno de estos identificados en la paciente.
Adicionalmente, en esta paciente fue documentado tromboembolismo pulmonar crónico. La incidencia de este trastorno es elevada en pacientes con vasculitis sistémicas primarias9. Esta asociación ha sido más estudiada en pacientes con GPA; en el ensayo clínico The Wegener's granulomatosis Clinical Occurrence of Thrombosis Study (WeCLOT), con una población de 180 pacientes con GPA, la incidencia de eventos tromboembólicos venosos fue 20 veces más alta que en la población general10. Las razones no están identificadas.
La presencia de anticuerpos antifosfolípidos no es una característica inusual en pacientes con vasculitis. Una mayor frecuencia de anticardiolipinas y anti β2 glicoproteína1 a títulos bajos se ha encontrado en pacientes con GPA; sin embargo, no se ha demostrado una asociación causal de los anticuerpos antifosfolípidos con trombosis11.
Además de GPA, los anticuerpos antifosfolípidos han sido reportados en asociación con poliarteritis nodosa, arteritis de células gigantes, polimialgia reumática, arteritis de Takayasu, síndrome de Behçet, granulomatosis eosinofílica con poliangitis y PAM3.
El estudio de Rees et al.12, realizado en 144 pacientes con vasculitis sistémica primaria, reportó una prevalencia del 17% de anticuerpos antifosfolípidos; el 6% (9 pacientes) cumplía criterios para SAF, y de estos últimos, 4 pacientes tenían vasculitis de pequeño vaso asociada a ANCA (2 casos con GPA y 2 casos con granulomatosis eosinofílica con poliangitis). No se describió ningún caso de PAM, lo cual es consistente con una búsqueda realizada en diferentes bases de consulta.
La ausencia de capilaritis en el análisis histológico de espécimen pulmonar, tomado en la primera hospitalización de la paciente, puede ser explicada por una hemorragia alveolar difusa secundaria a tromboembolismo pulmonar. En este orden de ideas y en consideración de los antecedentes obstétricos de pérdidas gestacionales ocurridos antes de la semana10 de gestación, asociados a los criterios de laboratorio dados por resultado positivo para anticoagulante lúpico y anti β2 glicoproteína1 IgG a títulos altos, se confirmó el diagnóstico de SAF.
En conclusión, consideramos que el espectro clínico de los pacientes con PAM continúa siendo definido; las características adicionales observadas en nuestra paciente sugirieron un SAF asociado, el cual fue finalmente confirmado. Por lo tanto, monitorizar el desarrollo de anticuerpos antifosfolípidos en pacientes con vasculitis sistémica primaria y evidencia de trombosis o morbilidad obstétrica puede ser importante para determinar la necesidad de anticoagulación profiláctica para prevención de trombosis.
FinanciaciónNinguna.
Conflicto de interesesNo hay conflicto de intereses.
