





Presentamos el primer caso reportado en Colombia de síndrome de Blau manifestado en el adulto, asociado a una mutación de novo en el gen de dominio de oligomerización de unión a nucleótidos 2 (NOD2) en el contexto de un caso de fiebre de origen desconocido (FOD).
Resumen del casoPaciente femenina de 44 años que presenta un cuadro de aproximadamente cuatro años de evolución consistente en episodios cíclicos de fiebre cuantificada, de duración aproximada de 20 días, con intervalos de remisión de seis a ocho meses, acompañada de dolor abdominal generalizado, polimialgias, poliartralgias y malestar general. Como antecedentes refiere tuberculosis (TB) hepática tratada, uveítis anterior, aparición de eritema nodoso en miembros inferiores con remisión espontánea y un episodio de parálisis facial periférica. Durante los estudios etiológicos se documentaron granulomas hepáticos, los cuales fueron llevados a biopsia, donde se encontraron múltiples granulomas no caseificantes. Se realizaron niveles de enzima convertidora de angiotensina, los cuales se encontraron dentro de los límites normales, y moleculares y microbiológicos para TB en la biopsia, los cuales fueron negativos. Posteriormente, se consideró como sospecha diagnóstica un síndrome autoinflamatorio, dada la persistencia del cuadro, por lo que se hicieron estudios genéticos que detectaron una mutación heterocigota de novo en el gen NOD2, la cual se asocia al síndrome de Blau.
ConclusionesLos síndromes autoinflamatorios, a pesar de que se presenten mayormente en la infancia, no se deben descartar en los adultos. En nuestro país no se conocen caso del síndrome de Blau manifestado en la edad adulta, por lo que este reporte de caso nos servirá para dar conocimiento a la comunidad científica al respecto.
We present the first case reported in Colombia of Blau syndrome manifested in adults associated with a de novo mutation in the NOD2 gene in the context of a case of fever of unknown origin.
Case summary44-year-old female patient presenting with a condition of approximately 4 years of evolution consisting of cyclical episodes of quantified fever lasting approximately 20 days, with remission intervals of 6 to 8 months, accompanied by generalized abdominal pain, polymyalgia, polyarthralgia, and general discomfort. Her medical history included treated liver tuberculosis (TB), anterior uveitis, appearance of erythema nodosum in the lower limbs with spontaneous remission, and an episode of peripheral facial paralysis. During the aetiological studies, hepatic granulomas were documented, which were taken to biopsy where multiple non-caseating granulomas were found. Angiotensin-converting enzyme levels were measured, which were found within normal limits, and molecular and microbiological limits for TB in the biopsy were negative. Subsequently, an autoinflammatory syndrome was considered a suspected diagnostic diagnosis given the persistence of the condition, so genetic studies were performed where a de novo heterozygous mutation was detected in the NOD2 gene, which is associated with Blau syndrome.
ConclusionsAutoinflammatory syndromes, although they occur mostly in childhood, should not be ruled out in adults. In our country there are no known cases of Blau syndrome manifesting in adulthood, so this case report will help us inform the scientific community about it.
La fiebre de origen desconocido (FOD) es una entidad que se caracteriza por presentar fiebre cuantificada de más de tres semanas de evolución, con al menos una semana de estudios sin poder esclarecer una causa aparente1. Las principales causas de FOD en Colombia corresponden a enfermedades infecciosas, en primer lugar, seguidas de neoplasias, enfermedades autoinmunes y causas misceláneas2. Dentro de la FOD existe un grupo de entidades conocidas como síndromes autoinflamatorios, los cuales son un grupo de enfermedades poco frecuentes que se caracterizan por presentar una mutación genética que lleva a una disfunción del sistema inmune innato, principalmente a nivel de los monocitos, lo que conduce a manifestaciones clínicas como episodios de fiebre recurrente, alteraciones cutáneas, dolor toracoabdominal y linfadenopatía, entre otros3.
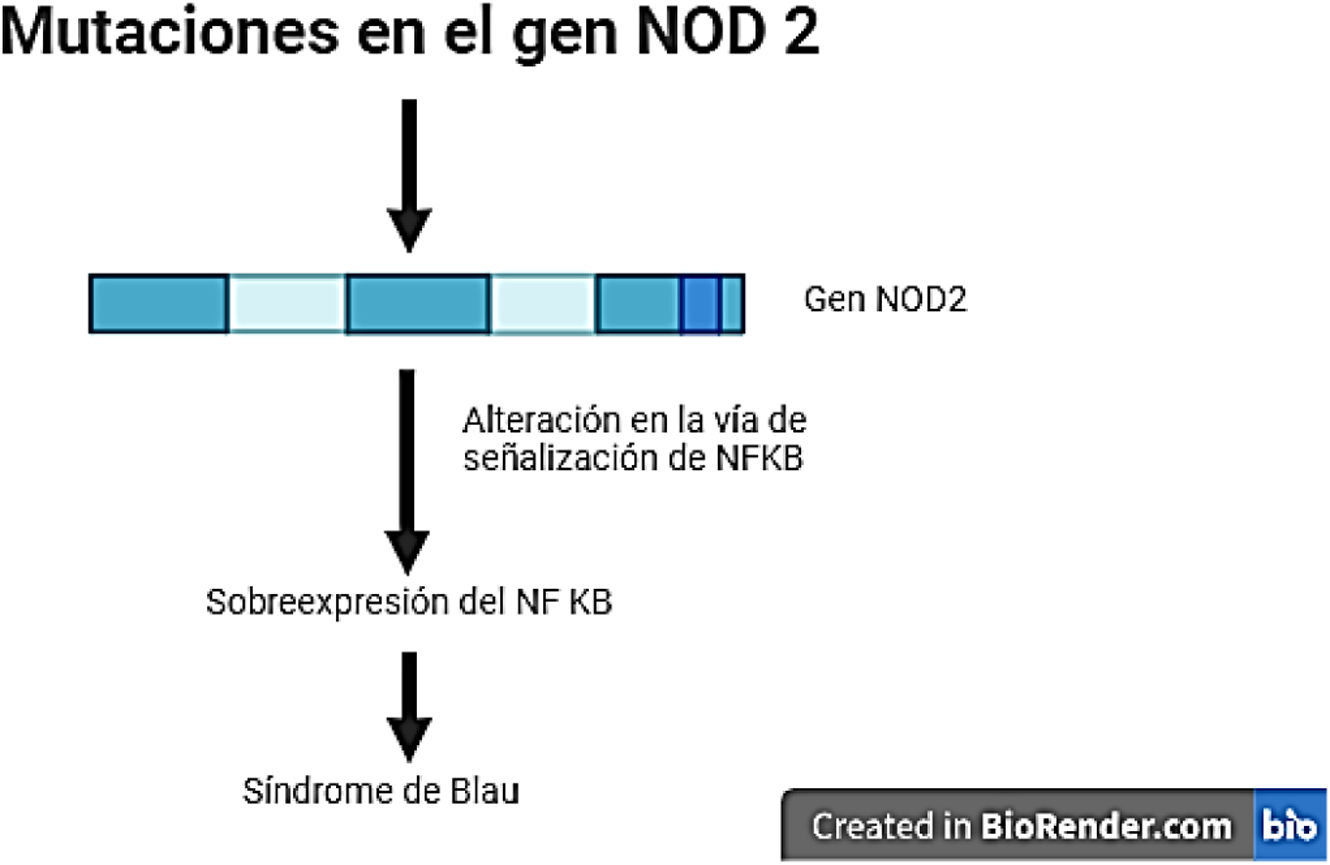
Entre los síndromes autoinflamatorios se encuentra el síndrome de Blau, una enfermedad autosómica dominante que se caracteriza por presentar episodios de fiebre cíclica asociada con una tríada clásica donde se presenta un compromiso granulomatoso asociado a manifestaciones articulares, cutáneas y oculares. Este síndrome suele manifestarse en los primeros cuatro años de vida y aparece en menos del 1% de los casos en la edad adulta4. Se asocia con una mutación del gen de dominio de oligomerización de unión a nucleótidos 2 (NOD2), el cual codifica para el receptor NOD2, involucrado en la respuesta inmune innata al activarse cuando entra en contacto con el lipopolisacárido de la pared celular bacteriana, lo que genera la activación del factor nuclear de cadenas ligeras kappa (NFkB). Esto último lleva a la producción de citocinas proinflamatorias y, por ende, a una reacción inflamatoria4,5. Una mutación en el gen NOD2 deriva en una sobreactivación del receptor del NFkB, lo que conlleva un aumento en la actividad de este, que a su vez genera una producción excesiva de citocinas proinflamatorias y un proceso inflamatorio desregulado5; es decir, la mutación con ganancia de función (GOF) del NOD2 en Blau, resulta en una hiperactivación del NFkB.
En el caso de nuestra paciente, al evidenciarse un compromiso granulomatoso a nivel hepático, se debían establecer como diagnósticos diferenciales enfermedades granulomatosas que pudieran estar asociadas a una mutación en el gen NOD2, por lo que se consideraron principalmente tuberculosis (TB), sarcoidosis, enfermedad de Crohn y el espectro de los síndromes autoinflamatorios.
Se valoró a una paciente de 44 años con un síndrome febril prolongado asociado a poliartralgias, dolor abdominal, malestar general, astenia y adinamia, con antecedente de uveítis, parálisis facial periférica y TB hepática tratada, en quien se documentó una inflamación granulomatosa crónica asociada con una mutación en el gen NOD2. La particularidad de este caso consiste en que es el primer reporte que se realiza en Colombia de un síndrome de Blau manifestado en la edad adulta.
Presentación del casoPaciente femenina de 44 años, natural, procedente y residente de Bogotá, de ocupación conductora de bus de transporte público, que ingresa a urgencias del Hospital Universitario Mayor Mederi con un cuadro clínico de dos semanas de evolución, consistente en picos febriles cuantificados hasta 39° C, de predominio nocturno, asociado a una sensación de escalofríos, diaforesis nocturna, polimialgias, poliartralgias y malestar general. Adicionalmente, refiere que hace aproximadamente cuatro años viene presentando dichos episodios de fiebre, la cual dura aproximadamente 20 días, con intervalos de remisión de seis a ocho meses. A la revisión por sistema, refirió dolor abdominal superior tipo cólico ocasional, no asociado a cambios en el hábito intestinal, artralgias ocasionales de predominio en manos, y muñecas sin rigidez matutina. Informó que un año atrás notó la aparición de lesiones eritematosas en la región pretibial de las piernas, bien delimitadas, brillantes y dolorosas, con desaparición espontánea. Como antecedentes patológicos refirió hipotiroidismo, TB hepática, para la cual recibió terapia con tetraconjugado por nueve meses; uveítis anterior asociada a desprendimiento de retina en 2019; una trombosis venosa profunda del miembro inferior derecho en 2018 anticoagulada con apixabán por ocho meses; y una parálisis facial periférica en 2018. Niega antecedentes familiares. Al examen físico de ingreso, como único hallazgo destacado se encontró una temperatura de 38,4° C, sin otras alteraciones de importancia.

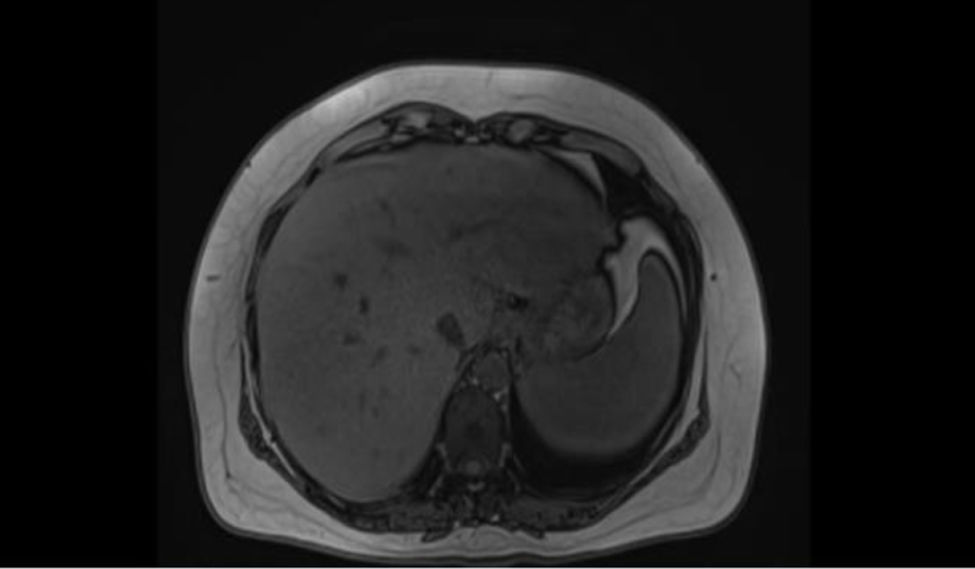
Resonancia magnética de abdomen con contraste, evidencia de múltiples lesiones redondeadas confluentes que comprometen en forma difusa todos los segmentos hepáticos, sin realce significativo posteriormente a la administración de contraste IV, compatibles con granulomas hepáticos.
IV: intravenoso.
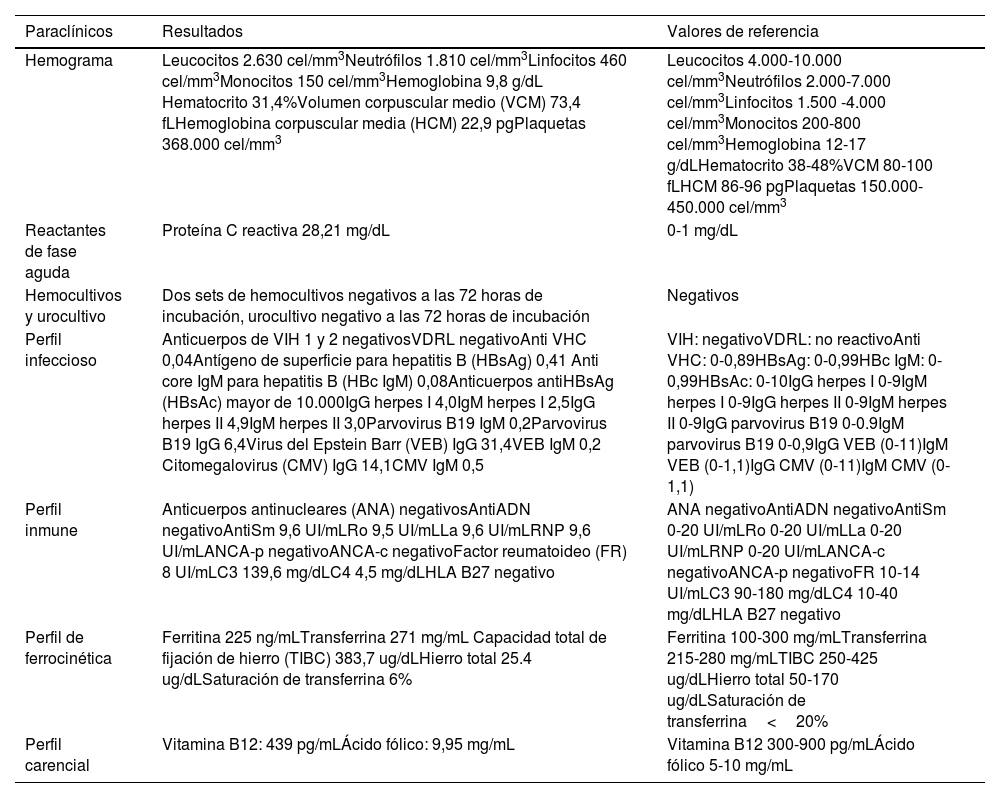
Inicialmente se consideró que la paciente estaba cursando con un cuadro de FOD clásica. Los estudios complementarios que se le realizaron se resumen en las tablas 1 y 2.
Resumen de paraclínicos
| Paraclínicos | Resultados | Valores de referencia |
|---|---|---|
| Hemograma | Leucocitos 2.630 cel/mm3Neutrófilos 1.810 cel/mm3Linfocitos 460 cel/mm3Monocitos 150 cel/mm3Hemoglobina 9,8 g/dL Hematocrito 31,4%Volumen corpuscular medio (VCM) 73,4 fLHemoglobina corpuscular media (HCM) 22,9 pgPlaquetas 368.000 cel/mm3 | Leucocitos 4.000-10.000 cel/mm3Neutrófilos 2.000-7.000 cel/mm3Linfocitos 1.500 -4.000 cel/mm3Monocitos 200-800 cel/mm3Hemoglobina 12-17 g/dLHematocrito 38-48%VCM 80-100 fLHCM 86-96 pgPlaquetas 150.000-450.000 cel/mm3 |
| Reactantes de fase aguda | Proteína C reactiva 28,21 mg/dL | 0-1 mg/dL |
| Hemocultivos y urocultivo | Dos sets de hemocultivos negativos a las 72 horas de incubación, urocultivo negativo a las 72 horas de incubación | Negativos |
| Perfil infeccioso | Anticuerpos de VIH 1 y 2 negativosVDRL negativoAnti VHC 0,04Antígeno de superficie para hepatitis B (HBsAg) 0,41 Anti core IgM para hepatitis B (HBc IgM) 0,08Anticuerpos antiHBsAg (HBsAc) mayor de 10.000IgG herpes I 4,0IgM herpes I 2,5IgG herpes II 4,9IgM herpes II 3,0Parvovirus B19 IgM 0,2Parvovirus B19 IgG 6,4Virus del Epstein Barr (VEB) IgG 31,4VEB IgM 0,2 Citomegalovirus (CMV) IgG 14,1CMV IgM 0,5 | VIH: negativoVDRL: no reactivoAnti VHC: 0-0,89HBsAg: 0-0,99HBc IgM: 0-0,99HBsAc: 0-10IgG herpes I 0-9IgM herpes I 0-9IgG herpes II 0-9IgM herpes II 0-9IgG parvovirus B19 0-0.9IgM parvovirus B19 0-0,9IgG VEB (0-11)IgM VEB (0-1,1)IgG CMV (0-11)IgM CMV (0-1,1) |
| Perfil inmune | Anticuerpos antinucleares (ANA) negativosAntiADN negativoAntiSm 9,6 UI/mLRo 9,5 UI/mLLa 9,6 UI/mLRNP 9,6 UI/mLANCA-p negativoANCA-c negativoFactor reumatoideo (FR) 8 UI/mLC3 139,6 mg/dLC4 4,5 mg/dLHLA B27 negativo | ANA negativoAntiADN negativoAntiSm 0-20 UI/mLRo 0-20 UI/mLLa 0-20 UI/mLRNP 0-20 UI/mLANCA-c negativoANCA-p negativoFR 10-14 UI/mLC3 90-180 mg/dLC4 10-40 mg/dLHLA B27 negativo |
| Perfil de ferrocinética | Ferritina 225 ng/mLTransferrina 271 mg/mL Capacidad total de fijación de hierro (TIBC) 383,7 ug/dLHierro total 25.4 ug/dLSaturación de transferrina 6% | Ferritina 100-300 mg/mLTransferrina 215-280 mg/mLTIBC 250-425 ug/dLHierro total 50-170 ug/dLSaturación de transferrina<20% |
| Perfil carencial | Vitamina B12: 439 pg/mLÁcido fólico: 9,95 mg/mL | Vitamina B12 300-900 pg/mLÁcido fólico 5-10 mg/mL |
ANCA: anticuerpos frente al citoplasma de los neutrófilos; HLA: antígenos leucocitarios humanos; Ig: inmunoglobulina; RNP: anticuerpos anti ribonucleoproteína nuclear; VDRL: prueba no treponémica: VHC: anticuerpos de hepatitis C; VIH: virus de la inmunodeficiencia humana.
Resumen de imágenes diagnósticas
| Imágenes | Resultados |
|---|---|
| Ecocardiograma TT | FEVI del 54%, sin disfunción sistólica ni diastólica; ausencia de alteraciones valvulares ni vegetaciones |
| Radiografía de articulaciones sacroilíacas | Estudio dentro de límites normales |
| Tomografía computarizada de cráneo simple | Estudio dentro de límites normales |
| Tomografía de cuello | Adenomegalia supraclavicular izquierda de 11 mm |
| Tomografía de alta resolución de tórax | Estudio dentro de límites normales |
| Resonancia magnética de abdomen con contraste (fig. 1) | Múltiples lesiones redondeadas, confluentes que comprometen en forma difusa todos los segmentos hepáticos, isointensas en T1, hiperintensas en T2 y difusión, sin realce significativo, posterior a administración de contraste IV, sugestivos de granulomas hepáticos |
FEVI: fracción de eyección del ventrículo izquierdo; IV: intravenoso; TT: transtorácico.
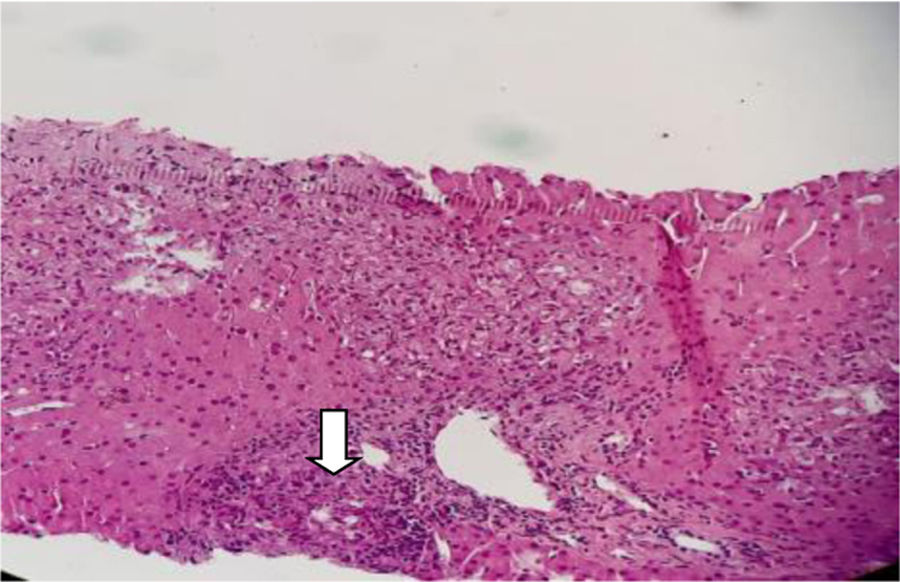
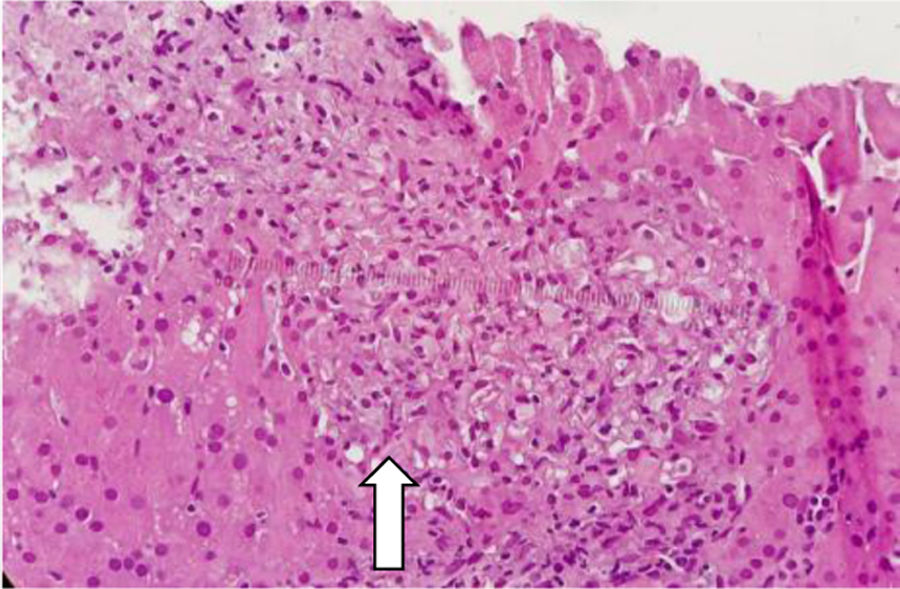
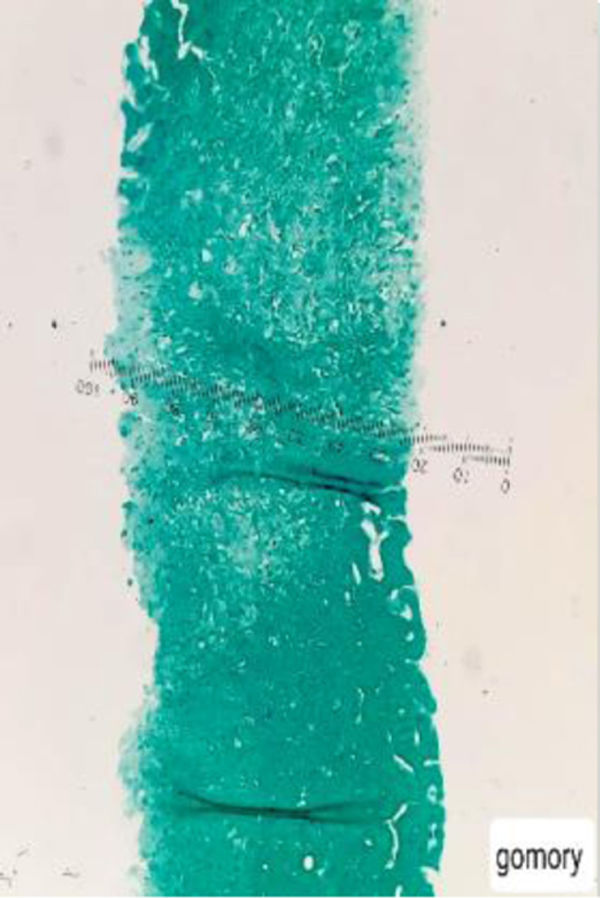
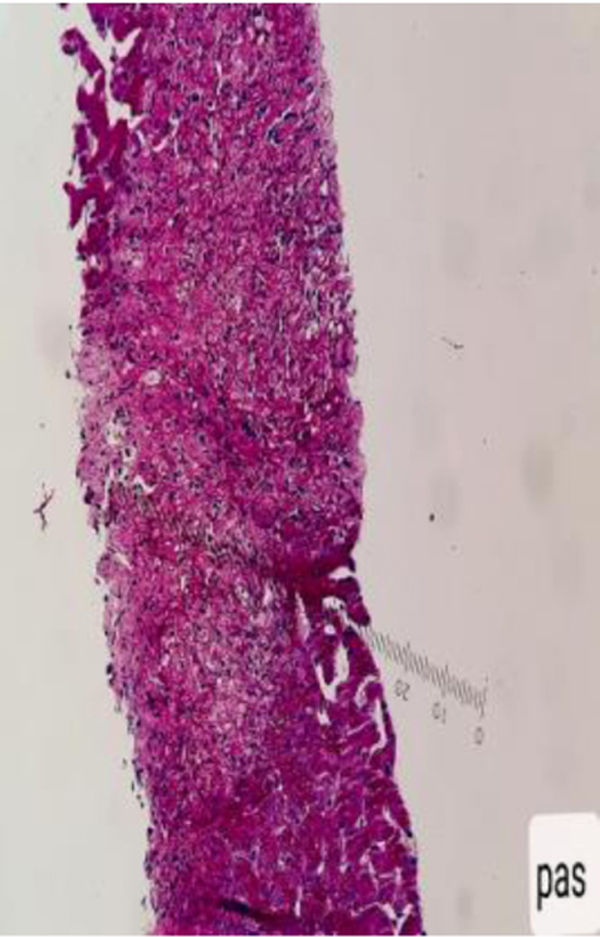
Con base en los hallazgos descritos, se consideró que la FOD de la paciente era secundaria a una enfermedad con compromiso inflamatorio y granulomatoso, teniendo como principal sospecha diagnóstica TB, dado el antecedente de la paciente de TB hepática. La paciente fue llevada a una biopsia hepática, la cual se describe en las figuras 2 y 3, con estudio de tinciones especiales, las cuales se pueden evidenciar en las figuras 4-6. Adicionalmente, como diferenciales de enfermedades con compromiso granulomatoso se solicitaron estudios específicos para sarcoidosis, TB hepática, colangitis biliar primaria y hepatitis autoinmune, los cuales se resumen en la tabla 3.
.")
 compuesto por acúmulos de histiocitos y macrófagos rodeados de células epitelioides, compatibles con una hepatitis granulomatosa.")
 negativo en donde se evidencia integralidad de los ductos biliares a nivel del parénquima hepático.")

 en biopsia hepática la cual es negativa para infección por hongos.")
Estudios complementarios como diferencial de hepatitis granulomatosa
| Paraclínico | Resultado |
|---|---|
| Enzima convertidora de angiotensina | 54 U/L (valor de referencia 9-67 U/L) |
| Perfil inmune con compromiso hepático | Anticuerpos antimitocondriales negativosAnticuerpos antimúsculo liso negativos |
| Antígeno urinario para histoplasma | Negativo |
| Estudio moleculares y microbiológicos para tuberculosis | PCR y cultivo de biopsia hepática negativos |
PCR: proteína C reactiva.
Dados los resultados descritos previamente, se inicia la sospecha diagnóstica de que la paciente pudiera estar cursando con un síndrome autoinflamatorio, debido a los episodios de fiebre recurrente, los granulomas hepáticos no necrotizantes y los demás estudios de extensión negativos.
De acuerdo con lo mencionado en el párrafo precedente, se decidió llevar a la paciente a una secuenciación más variantes en número de copia, mediante secuenciación de nueva generación (next generation sequencing [NGS]) en genes relacionados con fiebres periódicas y condiciones asociadas, en el cual se evidenció una mutación heterocigota a nivel del gen NOD2, con variante missense c.841 C>T, lo que genera el reemplazo de una leucina por una fenilalanina. Con base en el hallazgo de esta mutación, se consideraron dos diagnósticos diferenciales principales: la enfermedad de Crohn y el síndrome de Blau, dado que estas dos son las principales enfermedades que se asocian con las mutaciones del gen NOD2. Como estudio de la enfermedad inflamatoria intestinal se llevaron a cabo análisis endoscópicos, los cuales no mostraron los hallazgos típicos compatibles con una enfermedad de Crohn, además de una biopsia intestinal en la cual se reportó una mucosa sin evidencia de procesos inflamatorios, descartándose la presencia de granulomas, displasia y malignidad; como único posible diagnóstico queda el síndrome de Blau.
Finalmente, la paciente fue llevada a una prueba terapéutica con colchicina y esteroides a bajas dosis y se obtuvo una adecuada respuesta al tratamiento. Se hizo seguimiento por un año, en el cual no se presentó una nueva sintomatología ni nuevas recaídas.
DiscusiónEl síndrome de Blau hace parte de un subgrupo de los síndromes autoinflamatorios en el cual se da una desregulación del NFkB secundaria a una mutación en el gen NOD2. Este último codifica para el receptor NOD2 que está involucrado en la respuesta inmune innata al activarse cuando entra en contacto con el lipopolisacárido de la pared celular bacteriana, lo que genera la activación del NFkB. Lo anterior lleva a la producción de citocinas proinflamatorias, como se muestra en la figura 7, y, por ende, a una reacción inflamatoria6. Una mutación en el gen NOD2 conduce a una sobreactivación del receptor, lo que asimismo lleva a un aumento en la actividad de NFkB que a su vez genera una producción excesiva de citocinas proinflamatorias y, en consecuencia, un proceso inflamatorio desregulado4,5. Por lo general, las mutaciones asociadas son de herencia autosómica dominante, sin embargo, en ciertos casos pueden llegar a ser esporádicas. La edad de presentación más frecuente es en menores de cinco años, siendo menos del 1% de los casos la presentación en adultos5,6.

Las mutaciones del NOD2 que más se han asociado al síndrome de Blau son la sustitución de arginina por triptófano en la posición 334 (R334Q), la R334W y una sustitución L469F. Sin embargo, se han reportado más de 10 mutaciones en distintas partes del gen, las cuales se han asociado al fenotipo característico del síndrome de Blau. Los distintos tipos de mutaciones no se relacionan con la severidad de la enfermedad7.
Se ha visto que la exposición a cierto tipo de microorganismos, sumada a la predisposición genética, puede desencadenar los síntomas. Los microorganismos más asociados han sido las micobacterias, la salmonela y los virus herpes. Los periodos de fiebre se caracterizan por durar entre 15 días y un mes y suelen remitir por meses7.
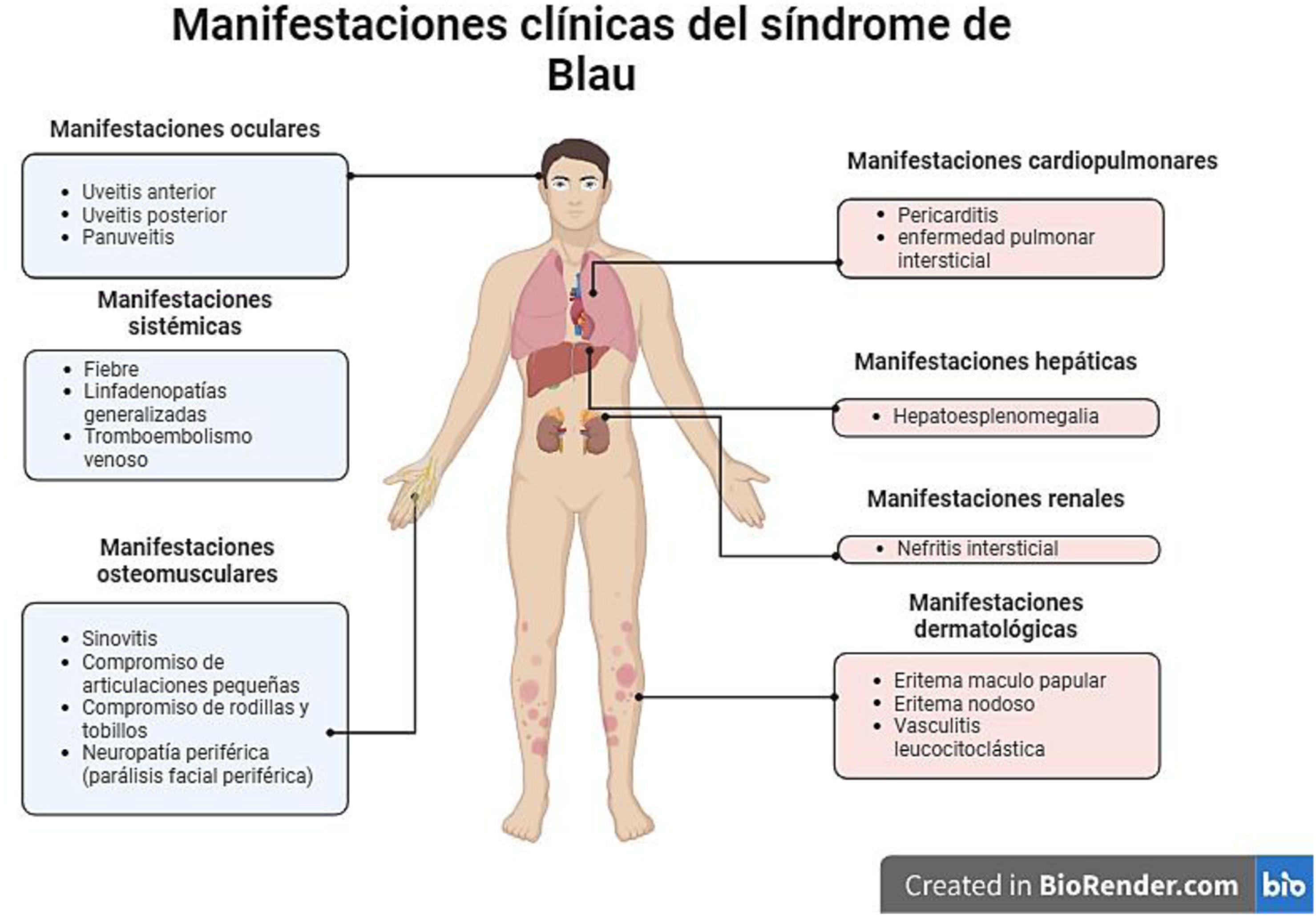
Los aspectos clínicos del síndrome de Blau se engloban en un grupo heterogéneo de manifestaciones que podemos visualizar de forma holística en la figura 8, sin embargo, uno de los aspectos fundamentales y más característicos en este síndrome es la presencia de granulomas no caseificantes en los tejidos afectados. De hecho, esta entidad es el único síndrome autoinflamatorio que se asocia con la presencia de granulomas inflamatorios. Estos se caracterizan por presentar un núcleo de macrófagos, células epiteliales, células gigantes multinucleadas y una corona de linfocitos con una presencia variable de fibroblastos8. Este hallazgo es concordante con los granulomas hepáticos documentados en la biopsia de nuestra paciente.

Entre los síntomas más frecuentes se encuentra el compromiso cutáneo, el cual se presenta en el 90% de los casos. Suele manifestarse en forma de un eritema maculopapular que compromete tronco y extremidades, o en forma de eritema nodoso. Esta manifestación suele ser la primera en presentarse y tiende a aparecer en el primer año de vida. En los estudios patológicos se pueden llegar a detectar granulomas no caseificantes7. En el caso de nuestra paciente, se presentó en forma de eritema nodoso.
En cuanto al compromiso articular, se presenta en el 95% de los casos. Suele manifestarse en forma de sinovitis articular, que compromete más que todo articulaciones pequeñas, principalmente las de las muñecas e interfalángicas proximales. También puede afectar las rodillas y los tobillos. Estas manifestaciones suelen iniciar entre los dos y los cuatro años de edad. No suele presentarse destrucción articular7. En este caso, dichas manifestaciones son concordantes con las referidas en la historia clínica de la paciente.
El compromiso ocular suele presentarse en el 60-80% de los casos y su principal manifestación clínica es la uveítis. Puede manifestarse como uveítis anterior, posterior o panuveítis. Usualmente es bilateral. De las tres manifestaciones típicas es la que se manifiesta de forma más tardía8. En el caso de nuestra paciente se presentó en forma de uveítis anterior, asociada a desprendimiento de retina.
Entre las manifestaciones atípicas podemos encontrar vasculitis leucocitoclástica, linfadenopatías generalizadas, esplenomegalia, neuropatía periférica (comprometiendo principalmente el nervio facial en forma de parálisis), nefritis intersticial, pericarditis, compromiso pulmonar intersticial y tromboembolismo venoso9. En el caso de nuestra paciente, llaman la atención los antecedentes de parálisis facial periférica y de tromboembolismo venoso, que son compatibles con esta enfermedad.
El diagnóstico del síndrome de Blau se basa en contar con un cuadro clínico compatible con la enfermedad, la detección de la mutación en el gen NOD2, ya sea esporádica o hereditaria, y una biopsia en la cual se confirme el compromiso granulomatoso no caseificante. El tratamiento se basa en la administración de colchicina y de corticoides a bajas dosis y por ciclos cortos8.
En el caso de la TB, esta se descartó, dado que no se hizo nunca un hallazgo microbiológico ni histopatológico a nivel de los granulomas hepáticos que pudiera confirmar a este microorganismo como agente causal del cuadro10.
Por otro lado, se descartó la enfermedad de Crohn, ya que los estudios imagenológicos, endoscópicos e histológicos no eran compatibles con dicho cuadro11.
Finalmente, no se consideró un cuadro de sarcoidosis, puesto que hasta el 90% de los pacientes tienen alteraciones pulmonares, ya sea la presencia de nódulos pulmonares o adenopatías hiliares, además de que no se detectaron alteraciones en los antígenos leucocitarios humanos (HLA), sumado a la enzima convertidora de angiotensina (ECA) en niveles normales12. Por otro lado, en ningún caso descrito de sarcoidosis en el adulto se han encontrado mutaciones asociadas al NOD213.
En el caso de nuestra paciente, se consideró que presentó una exposición a TB (ya que se obtuvo una prueba molecular positiva en esputo), la cual desencadenó las manifestaciones clínicas del síndrome de Blau. La paciente fue llevada a una prueba terapéutica con colchicina sumada a corticoides a bajas dosis, y se obtuvo una adecuada respuesta al tratamiento, sin embargo, al ser una entidad tan poco frecuente, no existen muchos datos de la efectividad de este tipo de tratamiento, sobre todo en adultos, ya que la poca evidencia que se tiene es en la edad pediátrica6. Con respecto al seguimiento de la paciente, este se ha realizado durante un año posterior al inicio del manejo con colchicina, con una adecuada respuesta clínica y ausencia de nuevos episodios febriles.
Por otro lado, se hace una búsqueda en las bases de datos genéticos como ClinVar, con el resultado de la variante de significado clínico incierto (VUS) en el gen NOD2, donde se refiere que la evidencia disponible es insuficiente para determinar el papel de esta variante en la enfermedad. ClinVar contiene una entrada para esta variante (ID de variación: 845687). Esta variante no se ha informado en la literatura en personas afectadas con enfermedades relacionadas con NOD2 ni está presente en las bases de datos de población (Genome Aggregation Database [gnomAD] sin frecuencia). Este cambio de secuencia reemplaza la leucina, que es neutra y no polar, por fenilalanina, que es neutra y no polar, en el codón 308 de la proteína NOD2 (p.Leu308Phe)14.
ConclusionesCasos como el que se reportó aquí, por lo general, no son de un abordaje sencillo, ya que suelen tener varios diagnósticos diferenciales. Cuando nos enfrentamos a casos como la FOD, se debe tener un amplio abanico diagnóstico, y se debe ser muy riguroso a la hora de evaluar las distintas posibles patologías. En nuestro caso, el diagnóstico diferencial de TB siempre estuvo presente, si bien existían otras posibilidades, las cuales finalmente se confirmaron, como fue llegar al síndrome de Blau. En Colombia no se conocen casos de este síndrome manifestados en la edad adulta, por lo que este reporte de caso nos servirá para poner en conocimiento a la comunidad científica de este.
FinanciaciónLos autores declaran que no recibieron financiación para la escritura o publicación de este artículo.
Consideraciones éticasCategoría de la investigación: según la Resolución 008430 de 1993, investigación sin riesgo. Dicho caso clínico se presentó en Hospital Universitario de Mederi Mayor en la cuidad de Bogotá, Colombia. En este caso no se requiere la obtención de consentimiento informado. La recolección de datos es de tipo secundario y se obtuvo a partir del registro de los datos consignados en la historia clínica. No se le identificará ni se tratarán aspectos sensitivos de la conducta del participante, ya que no existe ningún tipo de comunicación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
