




El síndrome antisintetasa corresponde a una enfermedad infrecuente, con grados de compromiso variable a nivel pulmonar, muscular, articular y cutáneo. Gracias a la introducción de nuevas pruebas diagnósticas, es posible detectar de forma más temprana la enfermedad y determinar la mejor estrategia de manejo. Sin embargo, en nuestro país la disponibilidad de estas pruebas es limitada. Se presenta el caso de una paciente con síndrome antisintetasa asociado con anticuerpos anti-PL7 en quien, gracias a la rápida identificación de dichos anticuerpos, se logró el inicio oportuno del tratamiento para la enfermedad.
Antisynthetase syndrome is a rare disease, with varying degrees of lung, muscle, joint, and skin involvement. Due to the introduction of new diagnostic tests, it is possible to detect the disease earlier and to determine the best treatment strategy. However, in our country the availability of these tests is limited. We present the case of a patient with antisynthetase syndrome associated with anti-PL7 antibodies in whom, thanks to the early identification of these antibodies, timely initiation of treatment for the disease was achieved.
El síndrome antisintetasa (SAS) es una enfermedad autoinmune infrecuente, con una presentación clínica heterogénea que comprende diferentes grados de compromiso a nivel muscular, pulmonar, articular y cutáneo1. Desde su descripción inicial hace 30 años2, la evidencia al respecto se ha incrementado. En la actualidad, se reconocen bastantes marcadores serológicos específicos adicionales a los anticuerpos anti-Jo1, que no solo permiten establecer el diagnóstico, sino predecir el comportamiento clínico3. Presentamos el caso de una paciente que inició con enfermedad pulmonar intersticial (EPI), manifestaciones cutáneas y compromiso muscular incipiente indicativo de miopatía inflamatoria (MI), en quien gracias a la identificación temprana del anticuerpo anti-PL7, el cual es específico para SAS, se logró una intervención oportuna de la enfermedad.
Presentación del casoMujer de 36 años, natural y procedente de Bogotá, valorada por nuestro servicio por primera vez en enero del 2021. Refirió inicio de síntomas en julio del 2020 con aparición de tos seca, intermitente, sin otros síntomas respiratorios asociados. Además, reportó aparición de máculas y pápulas eritematosas en el dorso de las manos sobre las articulaciones metacarpofalángicas e interfalángicas proximales, asociada con máculas eritematovioláceas no pruriginosas ni dolorosas en región periorbitaria y codos en septiembre del 2020. Asimismo, presentó sensación de debilidad muscular que se resolvió luego de 2semanas. Refería alopecia, aparición de úlceras orales intermitentes no dolorosas en paladar, disfagia para sólidos, artralgias de características inflamatorias en manos, carpo, codos y rodillas, asociado con rigidez matinal de 3h de duración y fenómeno de Raynaud (FR), todos los síntomas del mismo tiempo de evolución. En octubre del 2020 presentó exacerbación de la tos, acompañada de disnea que progresó desde clase funcional I/IV hasta III/IV, lo cual la llevó a requerir manejo hospitalario.
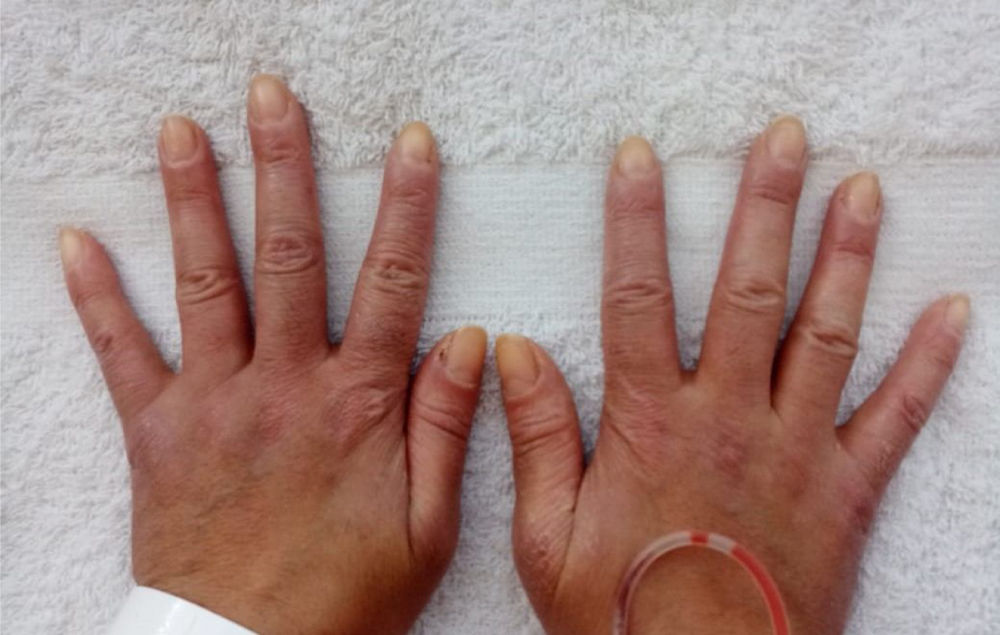
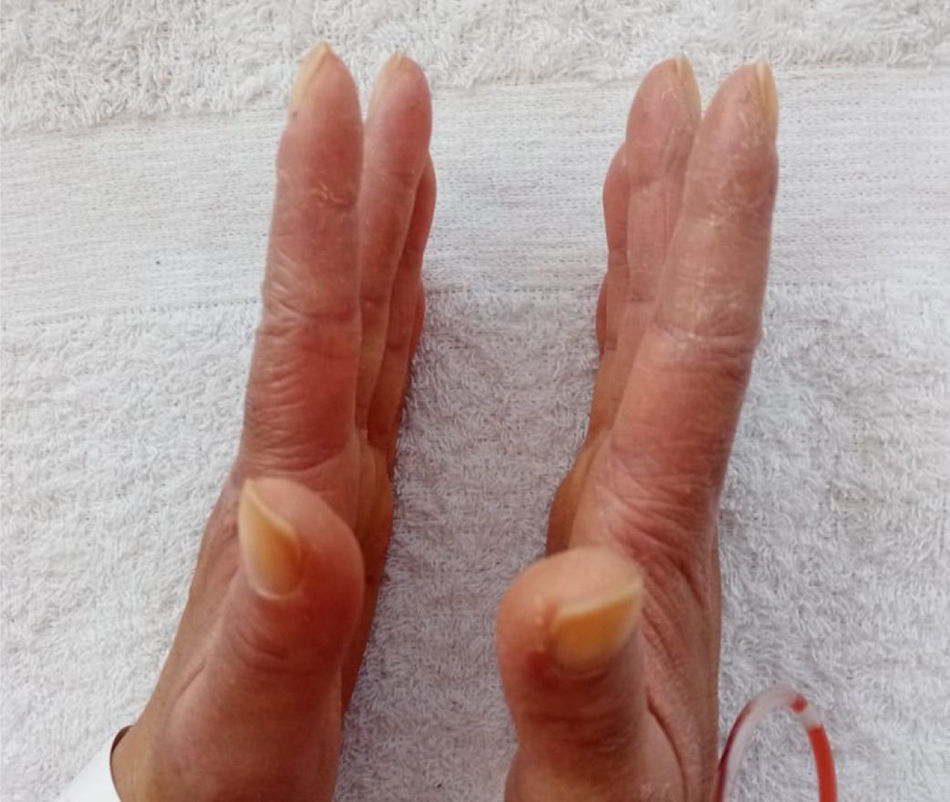
Estuvo hospitalizada en 2ocasiones previas a la valoración, la primera en octubre del 2020 por la presencia de los síntomas respiratorios descritos. Se descartó infección por COVID-19 y recibió manejo antibiótico por una semana con presunción diagnóstica de neumonía bacteriana. En la segunda oportunidad, en diciembre del 2020, debido a sintomatología similar, recibió manejo antibiótico por 2semanas y egresó con diagnóstico de EPI asociada a probable dermatomiositis, por lo que se encontraba recibiendo tratamiento con azatioprina 50mg cada 12 h, prednisolona 20mg/día y oxígeno suplementario por cánula nasal a 2 l/min las 24 h del día. Como antecedentes presentó infección por COVID-19 en junio del 2020, por lo cual requirió manejo ambulatorio. Refirió antecedente de ooforectomía izquierda por tumor de ovario de características inciertas. Su madre tiene antecedente de lupus eritematoso sistémico (LES). En el examen físico se documentó eritema en heliotropo, pápulas de Gottron, signo de Gottron y manos de mecánico (figs. 1 y 2). Se encontró además la presencia de FR, sin evidencia de sinovitis en la valoración. En la auscultación se advirtió la presencia de estertores en ambas bases pulmonares. No se encontraron alteraciones en el examen físico abdominal ni en el neurológico.


En los exámenes de laboratorio de diciembre del 2020 se evidenció: leucocitosis ligera, función renal normal, uroanálisis sin alteraciones, perfil lipídico normal, función tiroidea normal, leve elevación de la creatincinasa total y de la aldolasa, además de elevación de las transaminasas. La radiografía de tórax de octubre del 2020 presentaba opacidades en vidrio esmerilado de distribución periférica en ambos campos pulmonares. En diciembre del 2020 se realizó una tomografía de tórax (TACAR) con hallazgos de parches de vidrio esmerilado de distribución periférica y aleatoria en ambos campos pulmonares, con compromiso aproximado del 20% del parénquima pulmonar. El perfil inmune se presenta en la tabla 1. Durante su hospitalización de diciembre del 2020 se realizó un ecocardiograma transtorácico, sin alteraciones, en tanto que en la caminata de 6min se observó una caída de la saturación de oxígeno (SatO2) durante el ejercicio como único hallazgo patológico. La espirometría con curva flujo volumen encontró un volumen espirado forzado en el primer segundo (VEF1), con disminución de moderada a severa y capacidad vital forzada (CVF) normal.
Perfil inmune de la paciente durante diciembre del 2020
| Parámetro (ref.) | Valor reportado |
|---|---|
| CK-total (10-120 μg/l) | 330 μg/l |
| Aldolasa (1-7,6 U/l) | 9,4 U/l |
| Complemento C3 (90-180 mg/dl) | 145 mg/dl |
| Complemento C4 (10-40 mg/dl) | 32 mg/dl |
| ANA (IFI) | 1/160-moteado fino |
| Anti-SM (menor de 20 UE/ml) | 2 UE/ml |
| Anti-Ro/SSA (menor de 20 UE/ml) | 50 UE/ml |
| Anti-La/SSB (menor de 20 UE/ml) | 2 UE/ml |
| Anti-RNP (menor de 20 UE/ml) | 2 UE/ml |
| Anticoagulante lúpico (prueba presuntiva) | Negativo |
| B2 glicoproteína I IgG (menor de 8 U/l) | 2 U/l |
| B2 glicoproteína I IgM (menor de 8 U/l) | 3 U/l |
| Anticardiolipinas IgG (menor de 12 U IgG-FL/ml) | 4 (U IgG-FL/ml) |
| Anticardiolipinas IgM (menor de 12 U IgG-FL/ml) | 3 (U IgG-FL/ml) |
| Factor reumatoide (menor de 15 UI/ml) | Menor de 15 UI/ml |
| Anti-CCP (menor de 5 UI/ml) | Menor de 5 UI/ml |
| Anti-SCL-70 | Negativo |
| Anti-Jo1 | Negativo |
ANA: anticuerpos antinucleares; anti-CCP: anticuerpos contra péptidos critrulinados; anti-La/SSB: anticuerpos anti-La/SSB; anti-RNP: anticuerpos anti-U1RNP; anti-Ro/SSA: anticuerpos anti Ro/SSA; anti-SCL-70: anticuerpos anti-SCL70; anti-SM: anticuerpos anti-smith, CK: creatincinasa; IFI: inmunofluorescencia indirecta; IgG: inmunoglobulina tipo G; IgM: inmunoglobulina tipo M.
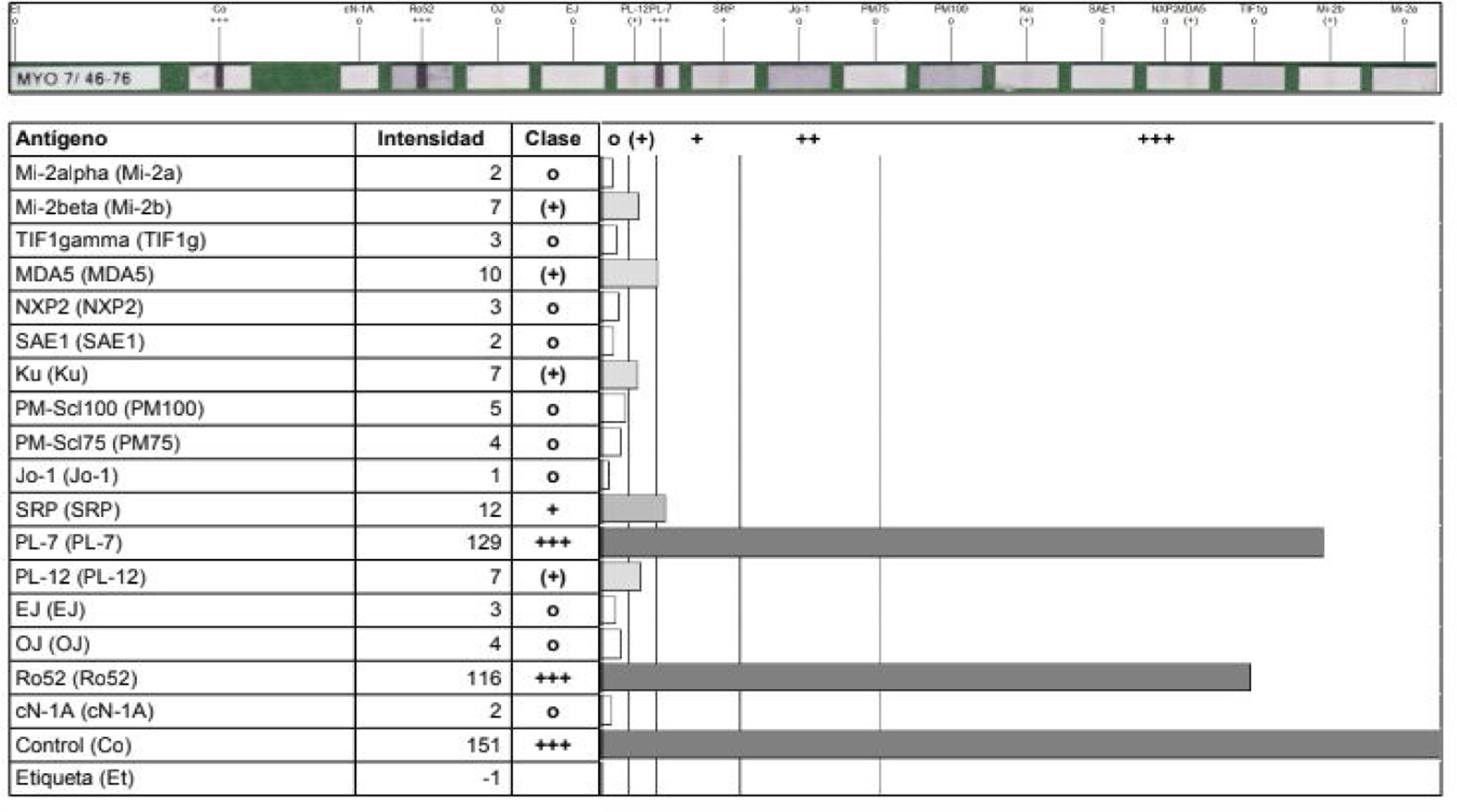
Todos estos hallazgos se interpretaron como probable caso de SAS, por lo que se realizó la medición de anticuerpos específicos (fig. 3), tras lo cual se confirmó el diagnóstico de SAS. Debido a los anticuerpos encontrados y al compromiso pulmonar de rápida progresión, se consideró enfermedad de mal pronóstico, por lo que se inició tratamiento con ciclofosfamida en bolos mensuales de 750mg por vía intravenosa y se suspendió la azatioprina. Se dio continuidad al tratamiento con prednisolona 20mg/día.

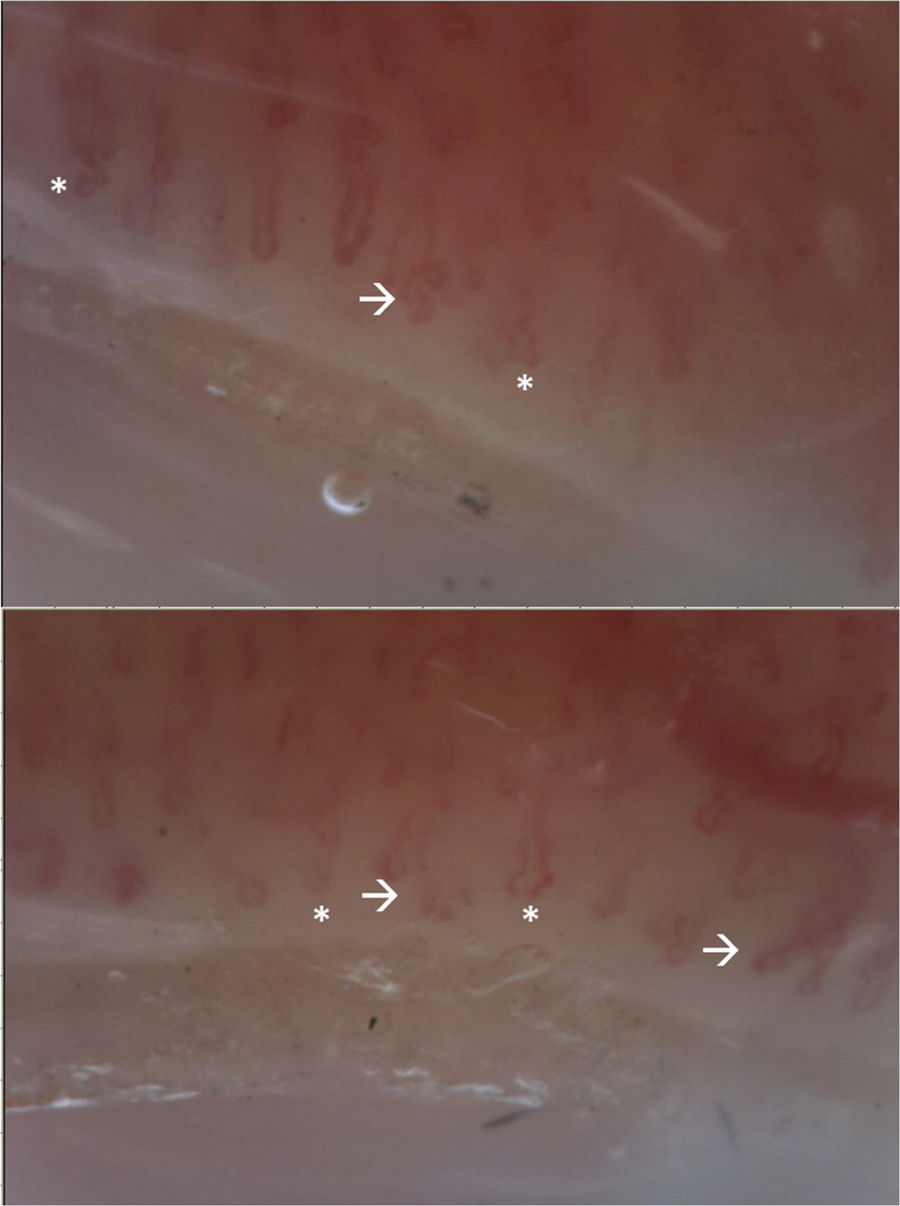
La paciente asistió a control en marzo del 2021 y recibió la primera dosis de ciclofosfamida sin complicaciones. Por la presencia de FR se realizó capilaroscopia con reporte de patrón no escleroderma (fig. 4). Además, se realizó: gammagrafía pulmonar normal, electromiografía y neuroconducción de las 4 extremidades —sin alteraciones— y ecografía transvaginal, que se situó dentro de límites normales. Se indicó continuar manejo con ciclofosfamida y prednisolona.
, sin microhemorragias ni megacapilares. Se evidencian ramificaciones (flecha). Patrón anormal, correspondiente a patrón no escleroderma. La presencia de múltiples ramificaciones sugiere la presencia de dermatomiositis.")
Video-capilaroscopia. Lecho vascular con edema marcado de papila, tortuosidades lineales y en su mismo eje (*), sin microhemorragias ni megacapilares. Se evidencian ramificaciones (flecha). Patrón anormal, correspondiente a patrón no escleroderma. La presencia de múltiples ramificaciones sugiere la presencia de dermatomiositis.
En abril del 2021 la paciente recibió la segunda dosis de ciclofosfamida sin que presentara complicaciones. Se realizó un control de pruebas de función pulmonar (PFP) que evidenció una capacidad de difusión de monóxido de carbono ajustada a la altura, moderadamente disminuida, con valor del 60%. La espirometría reportó una CVF del 69%, un VEF1 del 68% y una relación VEF1/CVF de 86. Se indicó continuar el mismo tratamiento, con seguimiento clínico mensual, sin embargo, por trámites administrativos con su aseguradora, asistió a control hasta junio del 2021, sin tratamiento farmacológico desde mayo de ese mismo año. Refería además deterioro de tos y disnea 20 días antes de la consulta, asociado con fiebre, mialgias, astenia y adinamia. Se documentó caída importante de la SatO2, además de sinovitis y FR, por lo que se hospitalizó para descartar proceso infeccioso activo. Se tomaron hemocultivos y urocultivo; con reportes negativos; se llevaron a cabo pruebas moleculares para SARS-CoV-2, con reportes negativos, además de baciloscopias negativas y un nuevo control de tomografía de tórax (fig. 5), en el que se documentó progresión de EPI. Se consideró sintomatología secundaria a reactivación de SAS, por lo que se reinició prednisolona a dosis de 20mg/día, se administró el tercer bolo de ciclofosfamida de forma intrahospitalaria y se dio egreso con orden del cuarto bolo de ciclofosfamida. Luego de esta cuarta dosis, la evolución fue estacionaria por lo que se indicó control de PFP y TACAR luego de completar esquema de tratamiento por 6meses con ciclofosfamida para determinar necesidad de inicio de rituximab.
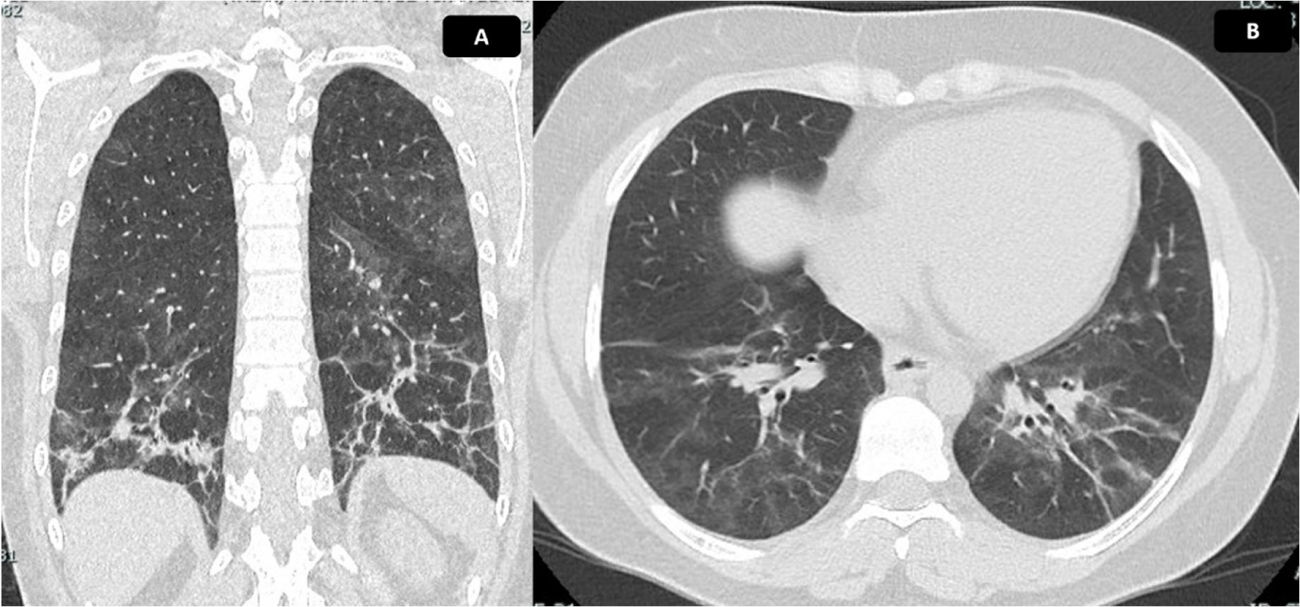
 y axial (B). Se evidencia vidrio esmerilado de distribución peribroncovascular, con mayor afectación de segmentos posteriores y lóbulos inferiores, donde se identifican reticulación, bandas parenquimatosas, engrosamiento de paredes bronquiales y bronquiectasias de tracción con pérdida de volumen asociada, debido a retracción de cisuras. Hallazgos compatibles con patrón de NINE fibrótica. Extensión calculada del 30%.")
Tomografía de tórax con hallazgos de EPI. Tomografía de tórax en corte coronal (A) y axial (B). Se evidencia vidrio esmerilado de distribución peribroncovascular, con mayor afectación de segmentos posteriores y lóbulos inferiores, donde se identifican reticulación, bandas parenquimatosas, engrosamiento de paredes bronquiales y bronquiectasias de tracción con pérdida de volumen asociada, debido a retracción de cisuras. Hallazgos compatibles con patrón de NINE fibrótica. Extensión calculada del 30%.
El SAS corresponde a una entidad clínica infrecuente que en la actualidad se reconoce como un subtipo de MI caracterizado por diferentes grados de compromiso muscular, pulmonar, articular y cutáneo4. Su patogénesis no se comprende completamente, sin embargo, cada vez se reconoce más la importancia de los anticuerpos dirigidos contra tRNA-sintetasas (AcSAS). Para el desarrollo de estos se requiere la interacción de factores genéticos y ambientales. Debido a factores mecánicos o por acción de microorganismos locales se genera lesión del endotelio pulmonar, lo que lleva a la exposición de autoantígenos que en individuos genéticamente predispuestos desencadenan fenómenos de pérdida de tolerancia y producción de autoanticuerpos5.
Se estima una prevalencia de 1-9/100.000 habitantes y puede corresponder hasta a un tercio de los casos de MI, siendo menos prevalente que la dermatomiositis y la polimiositis, pero más frecuente que la miopatía por cuerpos de inclusión y la miopatía necrosante inmunomediada6. Gracias a la introducción de nuevas técnicas diagnósticas y la identificación de nuevos AcSAS, es probable que en los próximos años estos valores cambien.
Se han propuesto diferentes criterios diagnósticos y, aunque hasta el momento ninguno es aceptado universalmente, todos reconocen la importancia de los autoanticuerpos para el diagnóstico de este síndrome7-9. Sin embargo, dada la heterogeneidad de la enfermedad, y que solo incluyen los anticuerpos anti-Jo1, se ha planteado la necesidad de establecer criterios clasificatorios claros para el SAS. En la actualidad se reconocen al menos 8 AcSAS, cada uno de ellos con particularidades especiales con respecto a sus manifestaciones clínicas, dentro de los que resaltan los anti-PL7 y anti-PL12, ya que confieren peor pronóstico a nivel pulmonar10.
El SAS asociado a anti-PL7 (SAS-PL7) se caracteriza por presentar de forma frecuente EPI severa, asociado con grados variables de compromiso muscular y cutáneo3,11,12. Cavagna et al. describieron las características clínicas de una cohorte de 95 pacientes con SAS-PL73 y encontraron que hasta el 76% presentaba EPI, siendo esta la característica clínica más común, además de la forma de inicio más frecuente de la enfermedad. El 49% presentó artritis en el curso de la enfermedad y hasta en el 11% se documentaron erosiones en radiografía convencional. El compromiso muscular estuvo presente en el 80%. Solo el 30% presentó esta manifestación al inicio de la enfermedad, sin embargo, el 61% desarrolló miositis en el curso de ella.
Con respecto al compromiso cutáneo, el 50% de los pacientes presentó FR y en el 42% se documentó la presencia de manos de mecánico. La mortalidad alcanzó el 13% en un seguimiento promedio de 93 meses3. A su vez, se ha descrito que el compromiso muscular suele ser leve y responder adecuadamente al tratamiento, por lo que puede ser clínicamente imperceptible, a diferencia del compromiso pulmonar, lo cual no excluye el diagnóstico de SAS-PL710,11. Jiang et al. encontraron además que la neumonía intersticial no específica es el tipo de EPI encontrado con mayor frecuencia en este grupo de pacientes (72,7% de los casos), presentándose como patrón principal o asociado a neumonía en organización12.
Desde su introducción, la identificación de anti-PL7ha desempeñado un papel importante dentro de los pacientes con SAS. Marie et al. compararon el perfil clínico de pacientes con SAS-PL7, SAS asociado a anti-Jo1 (SAS-Jo1) y anti-PL12 (SAS-PL12)13 y encontraron que los pacientes con SAS-Jo1 presentaron un mayor grado de compromiso muscular y articular al compararse con SAS-PL7 y SAS-PL12. A su vez, los pacientes con SAS-PL7 y SAS-PL12 presentaron de forma más frecuente EPI, con aparición más temprana y severa en el curso de la enfermedad al compararse con SAS-Jo113. Debido al impacto del compromiso pulmonar en los pacientes con SAS, la identificación de anti-PL7 y anti-PL12 impacta los desenlaces a largo plazo y conlleva un peor pronóstico al compararse con anti-Jo110,13.
No existe un consenso para el manejo del SAS y la evidencia al respecto deriva las más de las veces de estudios observacionales y del tratamiento de las MI14. La identificación temprana del subtipo de enfermedad permitirá determinar la mejor estrategia de tratamiento, cuya base es la combinación de diferentes medicamentos inmunosupresores15. Con relación a la EPI secundaria a SAS, la primera línea de tratamiento corresponde a los glucocorticoides asociados con medicamentos inmunosupresores como azatioprina, micofenolato, tacrolimús, ciclosporina, ciclofosfamida o rituximab16. Existen además reportes del uso de inmunoglobulina, plasmaféresis y trasplante pulmonar17,18. Es primordial identificar la severidad de la EPI y su grado de progresión. En el caso de la EPI rápidamente progresiva y de las formas agudas y subagudas asociadas con la presencia de factores de mal pronóstico, existe evidencia que sugiere que el uso de ciclofosfamida en combinación con glucocorticoides podría mejorar el compromiso muscular, además de las pruebas de función pulmonar, siendo bien tolerada por los pacientes19. Por otro lado, cuando se presenta EPI crónica progresiva, se recomienda el uso de glucocorticoides en combinación con otros inmunosupresores, como azatioprina, micofenolato o ciclosporina20. En el caso de EPI refractaria, existe evidencia del uso de rituximab, tofacitinib o plasmaféresis20,21.
Debido a los múltiples reportes y estudios observacionales del uso de rituximab en la EPI asociada con las enfermedades del tejido conectivo, se desarrolló el estudio Recital, un ensayo clínico aleatorizado que comparó el uso de rituximab con el uso de ciclofosfamida en el tratamiento de la EPI asociada con enfermedades del tejido conectivo, incluyendo SAS, y buscó definir su posible papel como terapia de primera línea22. En la actualidad se está a la espera de sus resultados finales.
En el caso de nuestra paciente, el curso clínico ha sido similar al reportado en la literatura. Además, gracias a la identificación temprana de los anticuerpos anti-PL7 se determinó la necesidad de iniciar inmunosupresión con ciclofosfamida, lo que ha permitido alcanzar una estabilización de la enfermedad. Sin embargo, teniendo en cuenta los factores de mal pronóstico con los que cuenta, especialmente la presencia de anti-PL7, se requiere un seguimiento estricto para determinar la necesidad de inicio de terapias de segunda línea como rituximab.
En la actualidad, los AcSAS corresponden a la principal herramienta paraclínica para realizar un diagnóstico oportuno e iniciar un tratamiento acorde con la condición de cada paciente. El presente caso nos ilustra sobre la importancia de contar con estas pruebas diagnósticas en nuestro medio, además de la necesidad de sensibilizar a neumólogos, dermatólogos y reumatólogos sobre la posibilidad de estar ante esta enfermedad en los casos de MI.
FinanciaciónEl presente reporte de caso no recibió ninguna financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses para la elaboración de este artículo.
Ninguno.
