




La enfermedad de Vogt Koyanagi Harada compromete múltiples órganos tales como ojos, meninges, oídos y piel. El curso progresivo de la enfermedad puede llevar a ceguera y cofosis. Se describe un caso de esta enfermedad en mujer hispana (mestiza) con alteraciones visuales, cefalalgia, tinnitus e hipoacusia a quien se le encuentra uveítis posterior con desprendimientos serosos de retina en ambos ojos y meningitis linfocitaria. El objetivo del presente estudio es, mediante una revisión de la literatura, actualizar la patogénesis inmunogenética, conocer las estrategias diagnósticas y el tratamiento apropiado.
Vogt Koyanagi Harada disease affects several parts of the body, such as eyes, meninges, ears, and skin. The progressive course of the disease can lead to blindness and deafness. The case is presented of a Hispanic woman (mixed-race) with visual alterations, headache, tinnitus, hearing loss, and posterior uveitis with serous detachments of the retina in both eyes, as well as lymphocytic meningitis. The aim of the present study is to review the literature, the diagnostic strategies, and the appropriate treatment, as well as to update the immunogenetic pathogenesis of the disease.
Paciente femenina de 34 años de ocupación higienista oral. Asistió a la consulta externa de oftalmología por un cuadro de 6 semanas de evolución consistente en ver distorsionadas y brillantes las líneas rectas de las señales viales y de noche no percibir con certeza la dirección de los autos en movimiento; adicionalmente, visión borrosa y miodesopsias. Asociado al cuadro anterior, la paciente refiere episodios de 3 semanas de evolución de dolor de cabeza, tinnitus persistente, hipoacusia y deterioro visual con compromiso de la visión de colores. Presentó, además, episodios aislados de «olvidos por segundos» que le ocasionaban bloqueos breves y transitorios en el curso del pensamiento y del lenguaje. Como antecedente relevante refirió historia familiar de glaucoma.
Al examen oftalmológico se encontró insuficiencia de convergencia y exotropía. Agudeza visual 20/20 ambos ojos (AO), sin embargo, manifestó distorsión del objeto visual con la visión central por AO y alteración de la visión de colores, mayor por ojo derecho (OD). Se identifica al examen hiperemia conjuntival bulbar en AO, córneas claras sin precipitados queráticos, Tyndall (−), iris y pupilas de aspecto sano, reflejos fotomotor y consensual normales en AO, sin defecto pupilar aferente. Presión intraocular de 12mmHg en AO. Fondo de ojo medios claros, relación disco óptico-excavación (copa) 0,6/0,6, el grosor del anillo neurorretiniano alrededor de las papilas ópticas (inferior, superior, nasal y temporal) conservado, vasos en bayoneta, ligera nasalización de los vasos, relación arteria-vena 2/3, máculas con disminución del brillo y presencia de pliegues maculares bilateral mayor en mácula OD con aparente membrana epimacular (pucker). La impresión diagnóstica inicial de oftalmología fue glaucoma, membrana epirretiniana y uveítis posterior. Se le ordenó angiografía fluoresceínica de retina, fotos de fondo de ojo, tomografía óptica coherente de discos ópticos y máculas, y campimetría 24-2 de AO. Por la cefalalgia y alteraciones cognitivas fue remitida a neurología donde, adicional a los hallazgos oftalmológicos descritos, se le encontró rigidez de nuca. Basados en lo anterior se realizó un diagnóstico sindromático:
- 1)
Síndrome de pares: ii par bilateral por compromiso maculorretiniano y viii par por compromiso auditivo bilateral.
- 2)
Síndrome meningoencefálico, de etiología a estudio.
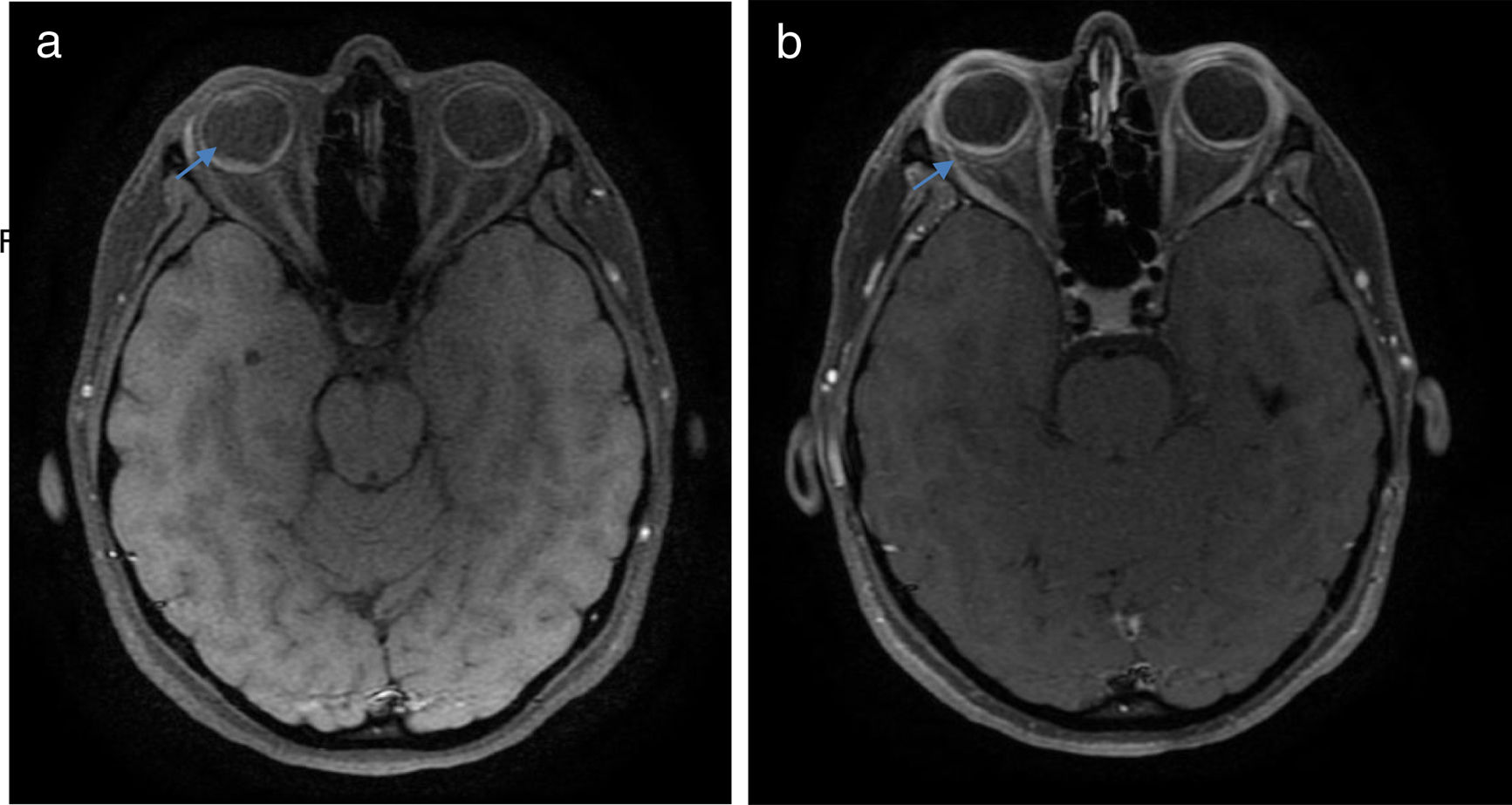
Una vez realizado el diagnóstico sindromático, se ordena hospitalización para estudio y tratamiento. Se solicitaron paraclínicos con resultados reportados dentro de los parámetros normales (tabla 1). La resonancia magnética (RM) de cerebro, simple y con contraste, sin alteraciones en parénquimas y sin realce meníngeo. En las órbitas, la RM mostró aumento del grosor de la coroides en ambos globos oculares con escleras íntegras, hallazgos compatibles con proceso ocular inflamatorio (fig. 1). La tomografía de coherencia óptica (OCT) de mácula (fig. 2 a) mostró engrosamiento coroideo y desprendimientos serosos de retina. La foto de fondo de ojo (fig. 3) y la angiografía digital con fluoresceína (fig. 4) demostraron lesiones uveorretinianas con compromiso del epitelio pigmentario y desprendimientos serosos de retina. Estudio de líquido cefalorraquídeo (LCR) obtenido por punción lumbar (PL) informó pleocitosis linfocitaria. La audiometría registró hipoacusia neurosensorial bilateral. Con base en la historia, los hallazgos clínicos, los resultados de laboratorio y en las imágenes, se hace diagnóstico definitivo de sindromático uveomeningoencefálico por la enfermedad de Vogt Koyanagi Harada (VKH).
Laboratorios
| Hemoglobina: 14,50 g/dl |
| Hematocrito: 44% |
| Recuento leucocitario normal |
| Velocidad de sedimentación (VSG) 15,00 mm/ hora (normal hasta 25 mm/hora) |
| Glucosa: 80 mg/dl |
| Anticuerpos antinucleares (ANA) (método IFI): 1/80 |
| TSH: 2,75 mU/ml (0,27-4,20) |
| Tiroxina T4 libre: 0,89 ng/dl |
| Creatinina: 0,80 mg/dl |
| Nitrógeno ureico (BUN): 20,3 mg/dl |
| Proteína C reactiva: 3,00 mg/l (normal 0-5 mg/l) |
| VDRL serología: no reactiva |
| Sodio: 138 meq/l |
| Potasio 4,01 meq/l |
| ENA: negativo |
| Transaminasa pirúvica (GPT-ALT): 14 U/L |
| Transaminasa oxalacética (GOT-AST): 18 U/L |
| Parcial de orina: normal |
| Anticuerpos IgG citomegalovirus (método electroquimioluminiscencia): 348,90 U/ml |
| Anticuerpos IgM citomegalovirus (método ELFA): 0,22 índice negativo menor a 0,7 |
| Virus de Epstein Barr IgG (método ELFA): 1,860 |
| Virus de Epstein Barr IgM (método ELFA): 0,010 índice negativo menor o igual a 0,11 |
 y luego (b) del contraste, con énfasis en órbitas, las cuales muestran aumento del espesor de la coroides, con realce difuso luego del contraste (flechas).")
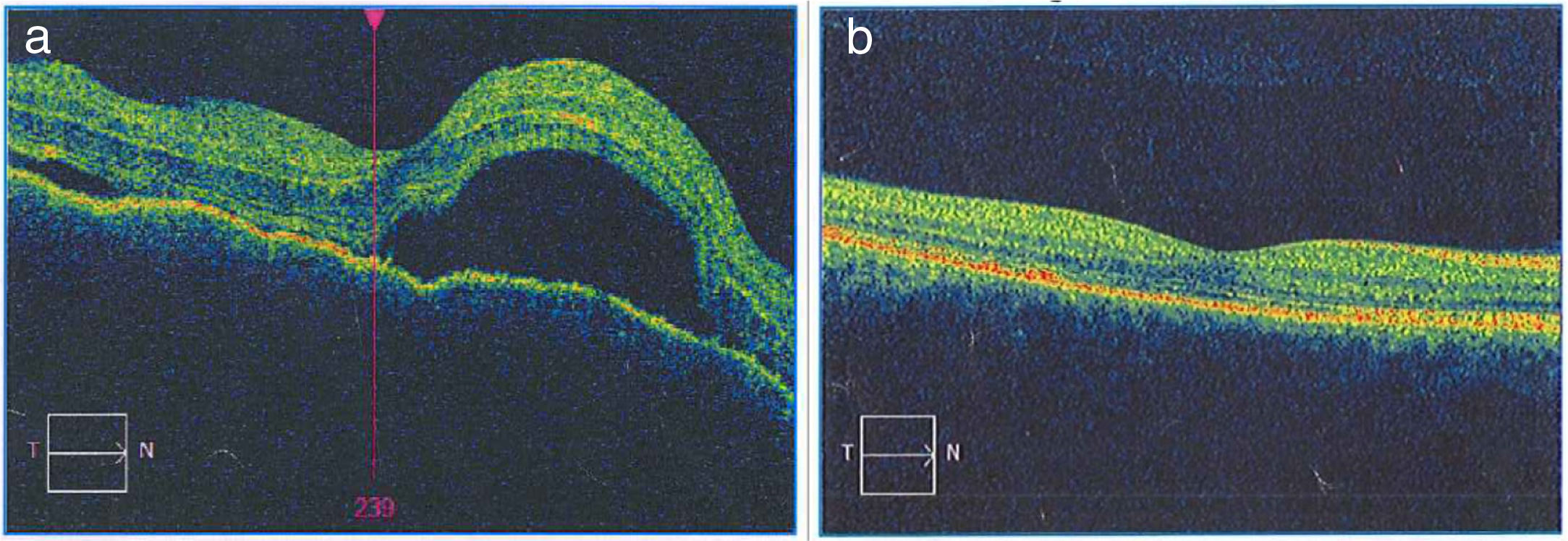
 La imagen de la OCT macular del OD diagnóstica permite observar el neuroepitelio con superficie irregular, pliegues retinianos en la membrana limitante interna, pérdida de la depresión foveal con aumento generalizado del espesor macular. También se observan múltiples desprendimientos serosos del neuroepitelio con acúmulo de fluido subretinal, imágenes que confirman la enfermedad de Vogt Koyanagi Harada. b) En la OCT macular del OD tomada 6 meses después de iniciado el tratamiento, se aprecia claramente resolución de los desprendimientos retinianos y el complejo EPR-membrana de Bruch-coriocapilares sin alteraciones. La coroides se muestra con patrón y grosor de apariencia normal.")
a) La imagen de la OCT macular del OD diagnóstica permite observar el neuroepitelio con superficie irregular, pliegues retinianos en la membrana limitante interna, pérdida de la depresión foveal con aumento generalizado del espesor macular. También se observan múltiples desprendimientos serosos del neuroepitelio con acúmulo de fluido subretinal, imágenes que confirman la enfermedad de Vogt Koyanagi Harada. b) En la OCT macular del OD tomada 6 meses después de iniciado el tratamiento, se aprecia claramente resolución de los desprendimientos retinianos y el complejo EPR-membrana de Bruch-coriocapilares sin alteraciones. La coroides se muestra con patrón y grosor de apariencia normal.

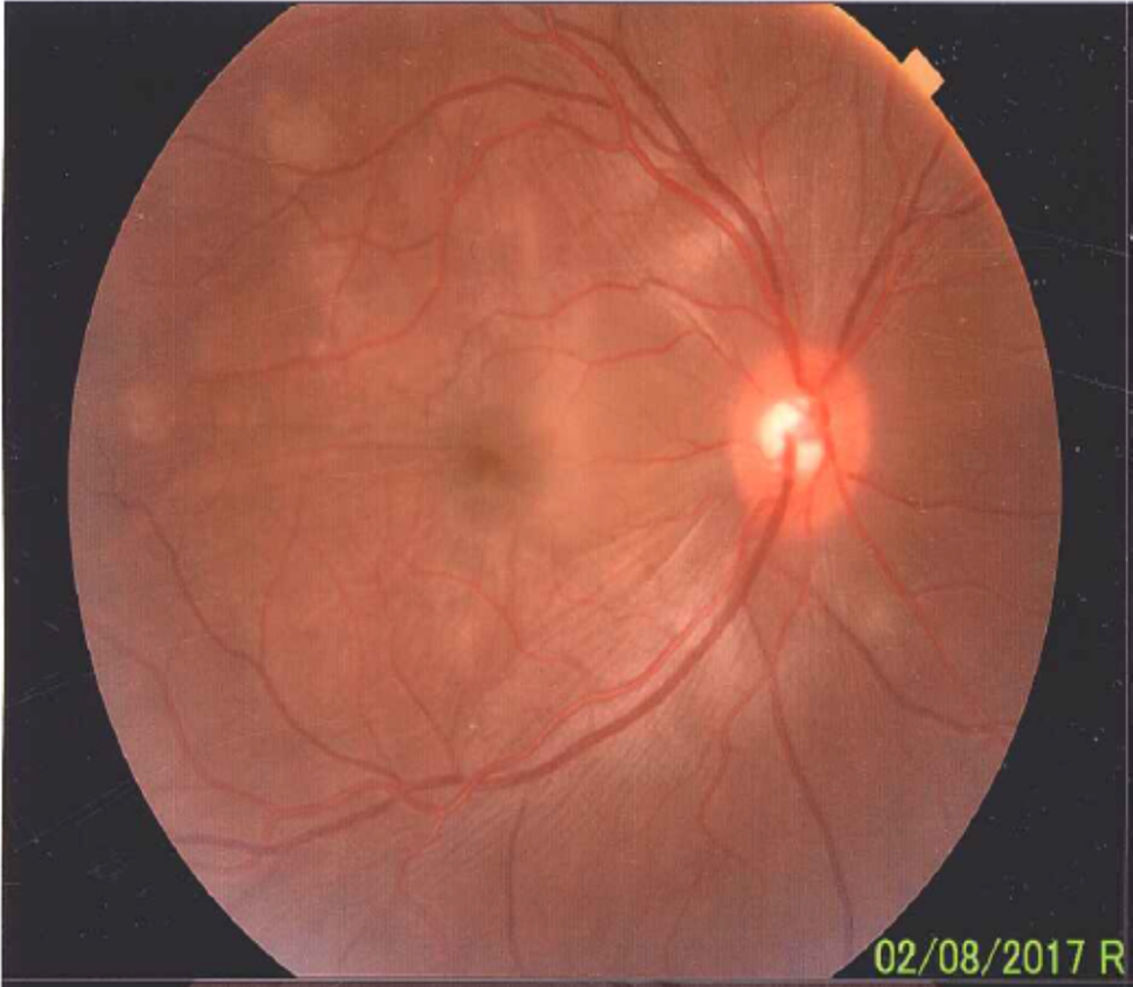
En el fondo de ojo se puede observar relación arteria-vena conservada, disco de bordes bien definidos, color amarillo rosado, excavación 0,4, emergencia y distribución vascular normal. El polo posterior, área macular y región peridiscal muestran pliegues en la membrana limitante interna y defectos difusos del amarillo en parches.

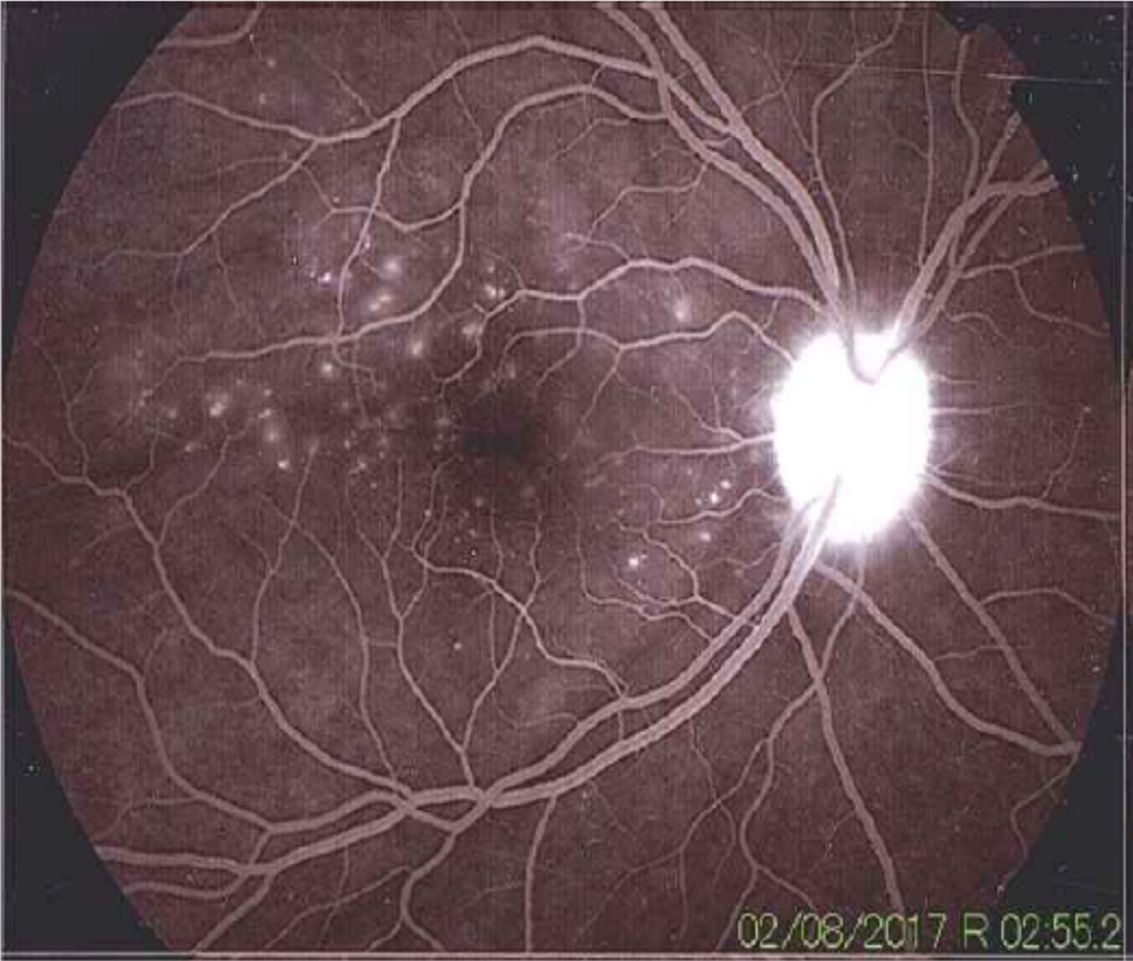
La angiografía mostró una dinámica circulatoria del medio adecuada con hiperfluorescencia del disco. En la foto del polo posterior se pueden observar múltiples defectos hiperfluorescentes por escape de colorante, sugestivos de desprendimientos serosos de retina. En el área macular también se muestran defectos hiperfluorescentes puntiformes.
Se inició tratamiento con bolos de metilprednisolona (1 g/día por 4 días), seguido por esquema de prednisolona (60mg/día y 10 días), después se agregó metotrexato (20mg/semana). La paciente presentó rápida mejoría clínica durante la primera semana de tratamiento: cedió la cefalalgia, el tinnitus y el meningismo. Valorada por retinólogo, ante presencia de edema macular y edema retiniano cistoide, 40 días después de iniciado el tratamiento, se decidió la aplicación intravítrea en OD de ranibizumab: una dosis y corticoide colirio OD por 7 días. Controles periódicos fueron demostrando mejoría progresiva, recuperación total de la agudeza visual y de la visión cromática, con preservación de su función visual luego de 12 meses de seguimiento. A partir del sexto mes de tratamiento se inició una reducción lenta y progresiva del corticoide oral. Al octavo mes de tratamiento presentó neuritis intercostal izquierda por virus varicela zóster que respondió bien a tratamiento con valciclovir. Persistió insuficiencia de convergencia y exoforia. La evaluación del tratamiento ha sido acompañada de imágenes OCT (fig. 2 b) que demuestran desaparición del edema retiniano y macular y retina bien aplicada, con reversión de los desprendimientos serosos retinianos a los 6 meses de tratamiento.
MetodologíaEl presente trabajo expone el caso de una paciente estudiada y tratada por la red prestadora de salud Colsanitas en Bogotá y se asocia a una revisión de la literatura. Para el desarrollo de la revisión y búsqueda de la literatura se partió de formular la pregunta cualitativa de investigación teniendo como guía, en su estructuración, la metodología PICO, sigla en donde «P» es la población objeto de estudio, «I» hace referencia a una forma de intervención médica, C es el «comparador» con otra forma de intervención y O procede del inglés «outcome» o desenlace.
En personas de mediana edad residentes en Colombia que consulten por presentar alteraciones oculares, miodesopsias, discromatopsias, dolor de cabeza, meningismo y sintomatología auditivo-vestibular (población, P), ¿es posible hacer el diagnóstico clínico de la enfermedad de VKH (intervención, I), o solo es posible hacer el diagnóstico de VKH mediante la práctica de estudios paraclínicos ordenados a partir de un diagnóstico sindromático (comparador, C), con el fin de iniciar el tratamiento oportunamente y con seguridad (outcome, O)?
Esta revisión encuentra justificación en la necesidad de obtener conocimiento actualizado de VKH, enfermedad rara y compleja, que muy ocasionalmente es reportada en Colombia. El propósito es generar conciencia sobre la importancia del diagnóstico rápido, el tratamiento oportuno con seguimiento especializado, tendiente a evitar las complicaciones severas que lo acompañan. El presente trabajo es una revisión narrativa de la literatura reciente sobre la enfermedad de VKH. Los artículos fueron seleccionados mediante la búsqueda en las bases de datos: Pubmed y Google Scholar. Los descriptores o palabras clave de búsqueda utilizados fueron en inglés: uveomeningoencephalitic syndrome and Harada; en español: síndrome uveomeningoencefálico y Harada.
Para la utilización correcta de la terminología se consultó la edición del 2017 de los Descriptores en Ciencias de la Salud en la página web:
Se realizó la investigación de evidencia disponible utilizando los descriptores de búsqueda. Como intervalo de búsqueda se decidió el lapso comprendido entre enero de 2010 y diciembre de 2017. Se identificaron en inglés 6.103 artículos potencialmente relevantes: 353 trabajos en Pubmed y 5.750 en Google Scholar. La revisión de los artículos fue realizada entre los meses de junio y diciembre de 2017 por 2 investigadores independientes.
Para la selección de los artículos se tuvo en cuenta que estuvieran publicados en el lapso señalado, la calidad metodológica, participación de autores expertos, así como la utilidad y relevancia de los contenidos de cara a la pregunta de investigación. Fueron seleccionados finalmente para la revisión 35 artículos que cumplieron los requisitos, 17 de Pubmed y 18 de Google Scholar. Con el fin de limitar el sesgo de selección, fueron implementadas estrategias como la búsqueda ordenada de los artículos suministrados por las bases de datos consultadas y la inclusión de trabajos obtenidos de la literatura gris. Uno de los artículos seleccionados para la revisión es anterior al lapso de búsqueda, porque se considera referencia obligada en razón a que contiene la revisión de criterios diagnósticos, aún vigentes, de la enfermedad de VKH.
Revisión de la literaturaAntecedentesLa enfermedad de VKH es un trastorno multisistémico caracterizado por inflamación ocular con síntomas y signos neurológicos, auditivos y dermatológicos. La enfermedad fue inicialmente descrita en 1906 por el oftalmólogo suizo Alfred Vogt, quien reportó el caso de un paciente con iridociclitis y poliosis. Yoshizo Koyanagi en un artículo de revisión, publicado en 1929, describió 16 casos en los que ilustra el curso de la enfermedad. Einosuke Harada, internista y oftalmólogo, además de los hallazgos extraoculares describe la coroiditis posterior aguda con desprendimientos exudativos de la retina y pleocitosis en el LCR1. Harada es quien logra hacer la descripción completa de la que se conoce hoy como la enfermedad de VKH.
EpidemiologíaLa enfermedad de VKH tiene una incidencia variable, más frecuente en personas del oriente asiático, hindúes, hispanos, mediterráneos e indígenas americanos; es infrecuente en caucásicos y suele afectar a pacientes entre los 20 y 50 años de edad, aunque también se presenta en niños1. Los estudios indican que las mujeres están más comprometidas que los hombres, en una proporción de 2 a 1, excepto en algunos estudios de Japón y China, que no mostraron diferencias en la prevalencia por género2.
Manifestaciones clínicasEl curso clínico de la enfermedad ha sido descrito por el comité internacional de expertos en VKH, convocado por la Sociedad Americana de Uveítis, en un artículo publicado en el 20013, donde se contemplan las 4 fases cronológicas clásicas: prodrómica, uveítis aguda, convaleciente y recurrente crónica.
Fase prodrómicaLos pacientes que desarrollan la fase prodrómica simulan un cuadro viral sistémico con claras manifestaciones neurológicas consistentes en cefalea, dolor orbitario, náuseas, hipoacusia o tinnitus, rigidez nucal y de espalda (meningismo) e hiperestesia de cuero cabelludo. Estas manifestaciones clínicas abarcan de 3 a 15 días antes de la fase de uveítis aguda1. En esta fase, rara vez se presentan signos focales cerebrales como confusión, afasia y hemiparesia.
El meningismo es el signo clínico extraocular más común, presente en el 49-67% de los pacientes con VKH4. La pleocitosis linfocítica en el LCR se encuentra en más del 80% de los pacientes y puede persistir hasta por 8 semanas3,5. Otros cambios en el LCR incluyen la presencia de macrófagos cargados de melanina específicos de VKH, aumento de proteínas y de la presión de apertura2. El análisis del LCR es una prueba importante para el diagnóstico precoz de VKH, particularmente en pacientes con dolor de cabeza, signos meníngeos claros y pocos signos oculares6; sin embargo, oftalmólogos expertos en uveítis, que trabajan en zonas geográficas endémicas de VKH y en centros especializados de referencia, afirman que raramente necesitan el estudio del LCR para el diagnóstico de enfermedad de VKH y plantean que la PL no debe ser un procedimiento rutinario, dado que los hallazgos clínicos y las avanzadas técnicas de imágenes en oftalmología, tales como la angiografía fluoresceínica y la OCT, confirman el diagnóstico clínico5,6.
La pérdida auditiva neurosensorial, el tinnitus, la plenitud auditiva y el vértigo suelen aparecer en la fase prodrómica y pueden ser concomitantes con uveítis activa7. Se demostró pérdida auditiva neurosensorial subjetiva en aproximadamente el 40% de los pacientes, mientras que el tinnitus fue reportado similarmente en el 38%8,9. Los síntomas vestibulares como vértigo y mareo se presentaron con menor frecuencia, descritos hasta en un 25% de los pacientes con VKH, es decir, en 1 de cada 410. El patrón de pérdida auditiva es bilateral y leve, principalmente en el rango de frecuencias altas9,10; sin embargo, se reportan casos de sordera súbita y severa bilateral11. Curiosamente, la pérdida auditiva detectada por audiometría es cercana al 90% de casos, significativamente mayor que la pérdida auditiva subjetiva (27%)9. Estos hallazgos audiológicos han permitido recomendar la audiometría como parte de la evaluación y control de la enfermedad.
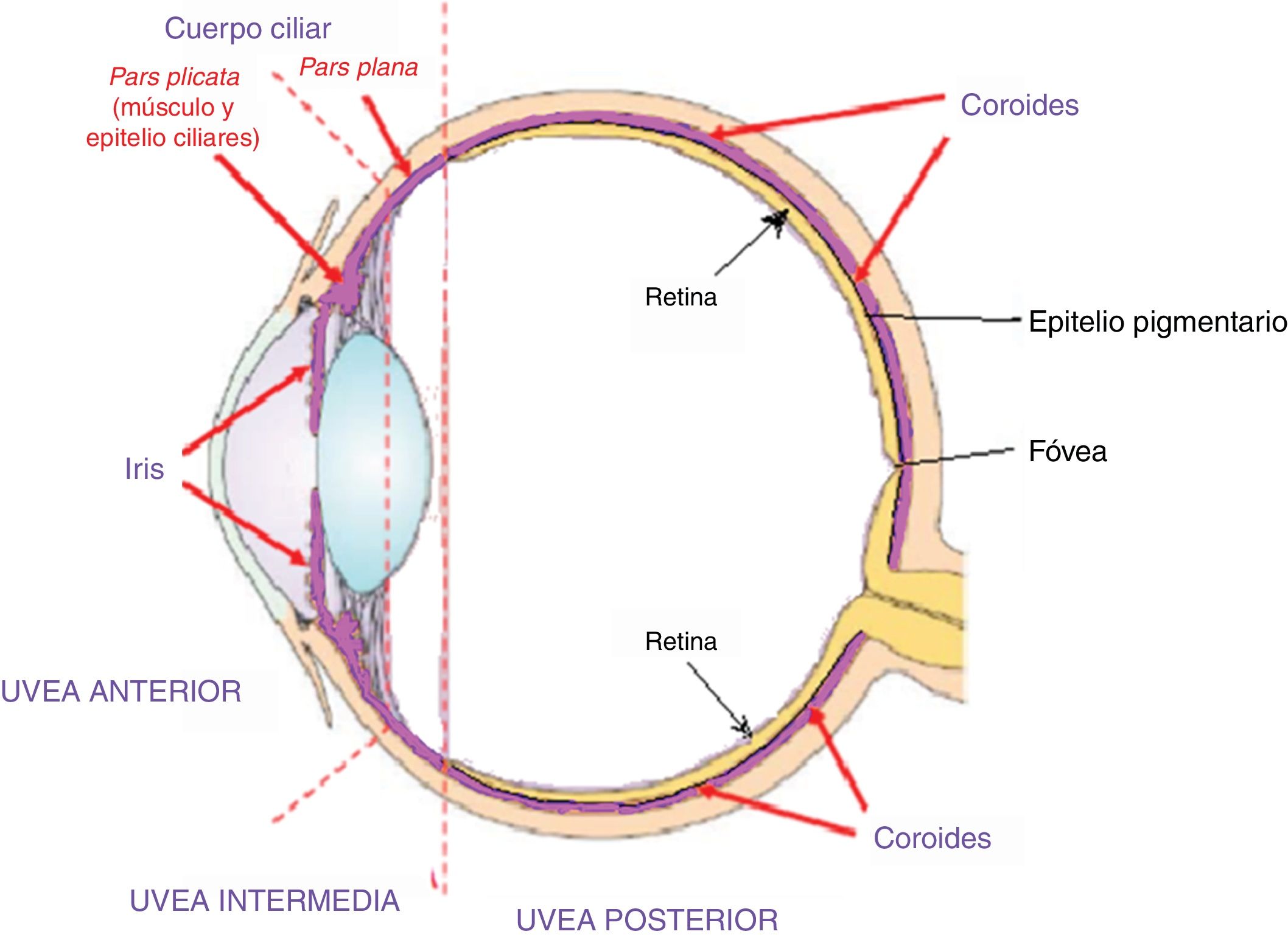
Fase uveítica agudaEsta se considera la fase distintiva de la enfermedad de VKH y puede prolongarse por varias semanas, en el curso de las cuales la mayoría de los pacientes consultan a oftalmología por presentar pérdida visual bilateral, usualmente asimétrica, debida a una coroiditis difusa. En la figura 5 se dibuja el tracto uveal en un corte transversal del globo ocular y se indica la coroides o úvea posterior inicialmente comprometida en VKH. La figura 6 es el dibujo de un segmento histológico de la unión del epitelio pigmentario de la retina (EPR) y la coroides, que son las estructuras especialmente afectadas por la inflamación autoinmune.
 que conforman la úvea anterior, la pars plana o úvea intermedia y la coroides o úvea posterior. Membrana limitante externa. Fuente: Tomado y modificado de https://www.google.com.co/search?q=tracto+uveal&source=lnms&tbm=isch&sa=X&ved=0ahUKEwi_uY3IiN_eAhXFx1kKHVpFDw4Q_AUIDigB&biw=1360&bih=626#imgrc=ydm7zLcwuBN8NM:.")
Este dibujo de corte transversal del ojo muestra el tracto uveal, resaltado en color violeta, con sus 4 componentes anatómicos: iris, cuerpo ciliar (pars plicata) que conforman la úvea anterior, la pars plana o úvea intermedia y la coroides o úvea posterior. Membrana limitante externa.
Fuente: Tomado y modificado de https://www.google.com.co/search?q=tracto+uveal&source=lnms&tbm=isch&sa=X&ved=0ahUKEwi_uY3IiN_eAhXFx1kKHVpFDw4Q_AUIDigB&biw=1360&bih=626#imgrc=ydm7zLcwuBN8NM:.

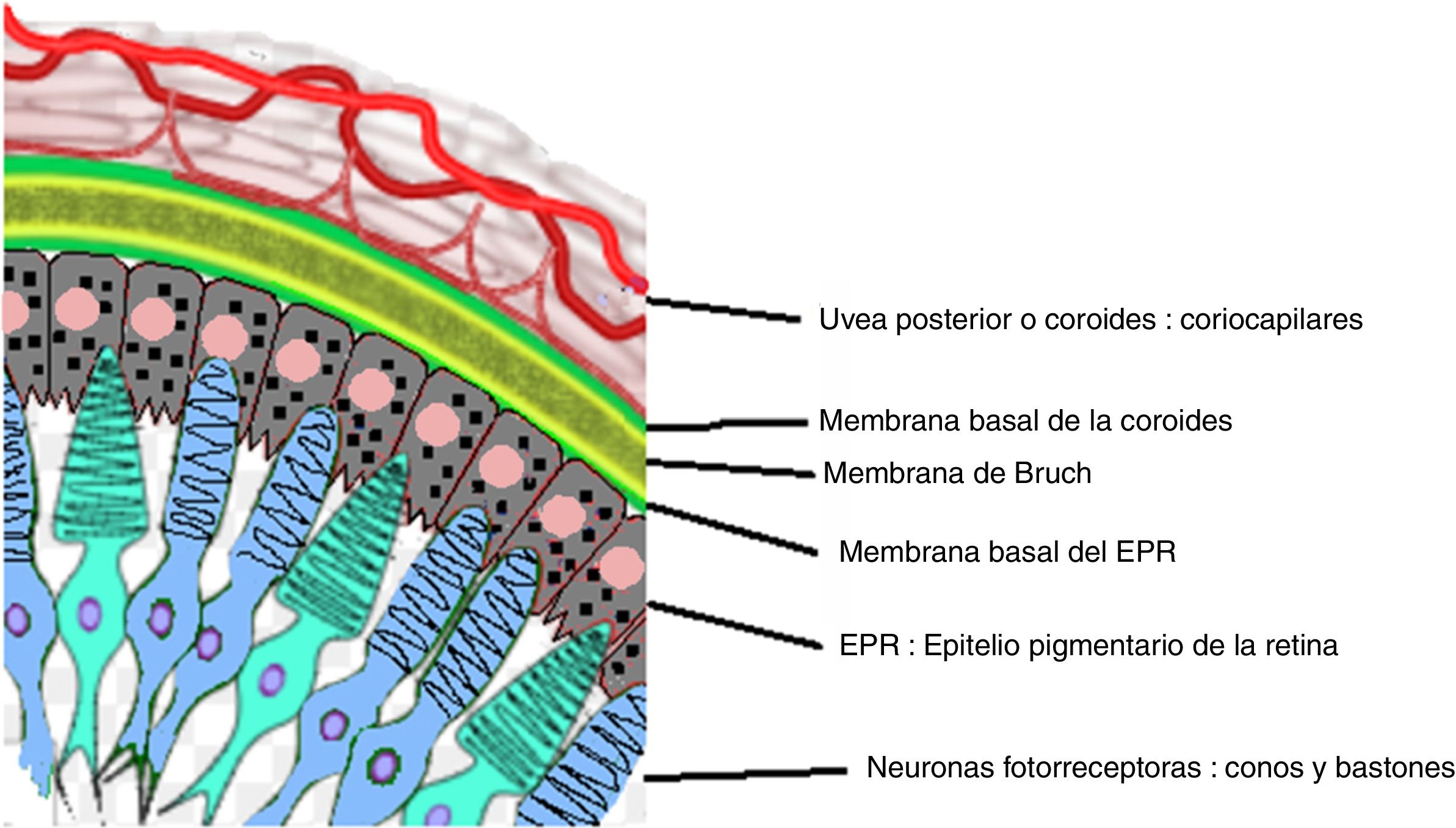
Dibujo que muestra la relación anatómica entre el entramado coriocapilar de la coroides y la capa más externa de la retina, el epitelio pigmentario. Estas estructuras son el blanco del compromiso inflamatorio ocular en la enfermedad de VKH.
Fuente: Tomado y modificado de https://en.wikipedia.org/wiki/Bruch%27s_membrane#/media/File:Buchs_membrane.svg.
En esta fase uveítica aguda el examinador puede observar edema e hiperemia del disco óptico antes de observar células en el vítreo y el clásico desprendimiento exudativo de retina bilateral5,6. El compromiso inflamatorio de la coroides altera la barrera hematorretiniana externa, conformada por la membrana de Bruch y el EPR, y provoca acumulación de fluido subretiniano y múltiples desprendimientos de retina, que pueden confluir y formar grandes desprendimientos ampollosos. La presencia de engrosamiento de la coroides de predominio peripapilar puede observarse claramente mediante una OCT12-14, hallazgos que se correlacionan con infiltración celular inflamatoria mononuclear difusa. En esta fase, la inflamación puede afectar la cámara anterior. Cuando se presenta edema del cuerpo ciliar, se puede elevar la presión intraocular y ocasionar glaucoma agudo por cierre del ángulo entre la base de inserción del iris y la córnea2,6.
Fase convalecienteLa fase convaleciente ocurre varias semanas después de la fase uveítica aguda, puede durar varios meses y se caracteriza por la aparición de zonas de despigmentación coroidea y cutánea. El signo de Sugiura o vitíligo perilímbico es el más precoz: puede aparecer en el primer mes de la enfermedad1,3. Con la pérdida de EPR y de melanocitos coroideos, el disco óptico pálido aparece sobre un fondo brillante rojo-anaranjado, color dado por la coroides despigmentada. Esta imagen evoca en el examinador el crepúsculo, con su característico «brillo o resplandor de la puesta del sol», y constituye el signo clínico conocido en la literatura inglesa como sunset glow fondo3, que aparece 2-3 meses tras la fase uveítica1. En esta fase, el oftalmólogo también puede observar focos de hiperpigmentación por migración de células del EPR, especialmente en pacientes hispanos, y múltiples lesiones pequeñas y redondeadas, blanco-amarillentas que corresponden histológicamente a los nódulos de Dalen-Fuchs6, que son agregados de linfocitos y macrófagos cargados de pigmento que aparecen hacia el polo posterior entre la membrana de Bruch y el EPR1.
La despigmentación de piel (vitíligo), pestañas, cejas y cabello (poliosis) y alopecia se observa principalmente durante esta fase, como un hallazgo característico3. Estas manifestaciones cutáneas son más comunes en los pacientes asiáticos5,15. El vitíligo suele distribuirse simétricamente y afectar la región facial, palpebral, tronco y zona cutánea sacra2.
Fase crónica recurrenteEn el curso de la fase convaleciente, después de meses de tratamiento, algunos pacientes pueden evolucionar hacia la fase crónica, que se caracteriza por la aparición de uveítis anterior granulomatosa recurrente, que lleva al desarrollo de nódulos en el iris, atrofia focal iridiana e hipotonía ocular1,4,7. Durante esta fase se ha informado de casos que han cursado con inflamación coroidea posterior, engrosamiento coroideo y desprendimientos de retina, demostrados por angiografía con verde de indocianina (AVI)1 y OCT16. Esta fase recurrente crónica suele aparecer en los 6 primeros meses de la enfermedad, como consecuencia de una disminución rápida o suspensión temprana de la corticoterapia16,17.
Derivadas de la inflamación intraocular crónica surgen complicaciones: las más frecuentes son las cataratas y el glaucoma, seguidas de neovascularización o fibrosis subretinal; con menos frecuencia, se presentan sinequias posteriores, neovascularización del disco óptico, anastomosis arteriovenosas y neovascularización coroidea4,6,7. Todas estas complicaciones comprometen severamente el pronóstico visual.
Etiología y patogénesisLa fisiopatología de la VKH ha sido ampliamente estudiada en las últimas 2 décadas; sin embargo, la etiología precisa no está aún bien establecida. Puede afirmarse que la patogénesis de la VKH es multifactorial, cuyo blanco principal es la capa coroidea del ojo (figs. 5 y 6). Aunque se han reportado etiologías inmunogenéticas y ambientales, el proceso inmunológico que conlleva a la enfermedad VKH es una respuesta autoinmune mediada por células T contra uno o más componentes antigénicos de los melanocitos en el ojo, oído, piel, plexos coroideos y cerebro1,7,18. Varios genes antígenos leucocitarios humanos (HLA) y no HLA han sido identificados y relacionados con la enfermedad VKH. Con base en esta diferenciación, se han clasificado los factores inmunogenéticos como asociados y no asociados al HLA18,19.
Los genes no HLA relacionados con la enfermedad son el IL-23A, IL-23R, IL-17F, IL-27. Estudios recientes en uveítis autoinmune experimental, en modelo animal clásico, señalan un papel central a las células Th 17 en la patogénesis de la uveítis20. Estos resultados en el modelo animal fueron confirmados en un estudio de pacientes con VKH, según el cual se demuestra que la producción de IL-17, citocina principal de células Th 17, es parte esencial del mecanismo involucrado en el desarrollo de la uveítis de los enfermos de VKH. Este estudio logra demostrar que la expresión reducida de IL-27, que es una citocina antiinflamatoria que regula la activación de linfocitos Th 17, resulta en una mayor expresión de Th 17 en pacientes VKH y uveítis activa20. La importancia de estos hallazgos radica en que la manipulación de IL-27 puede ofrecer nuevos tratamientos para VKH. Por ahora se sabe que el tratamiento con corticosteroides, al provocar una regulación positiva de IL-27 y una regulación negativa de IL-17, contribuyen en la resolución de la inflamación intraocular20.
También han sido identificados varios genes HLA que se relacionan con la enfermedad de VKH, pero la fuerza de asociación entre dichos genes y la enfermedad no es la misma en diferentes grupos étnicos. Así, por ejemplo, la asociación de VKH con el HDL-DR4/DRw53 fue encontrado en asiáticos, indígenas norteamericanos y en pacientes hispanos5,18. En cambio, los alelos HLA-DRB1*0405 y HLA-DRB1*0410 se asociaron fuertemente con VKH en pacientes hindúes4.
Además de las respuestas autoinmunes a los melanocitos, la enfermedad VKH parece estar causada por actividad autoinmune contra antígenos asociados a melanocitos. Se ha demostrado que antígenos de las proteínas de la familia de la tirosinasa y GP100, proteína expresada en la membrana basal del melanosoma, son blancos antigénicos reconocidos por células T de pacientes VKH con HLA DRB1*0405 positivo y han sido relacionados con la causa y la enfermedad de VKH5,18.
KU-MEL-1 es otro autoantígeno que se encuentra ampliamente expresado en la mayoría de las líneas celulares de melanoma, muestras de tejidos de melanoma y en melanocitos cultivados18. El anticuerpo contra KU-MEL-1 positivo en suero presentó una asociación muy significativa en los pacientes de VKH con HLA- DRB1*0405 positivo, en comparación con pacientes afectados con otras uveítis y con controles sanos5,18. Esta asociación implica una función primordial de linfocitos T CD4+ específicos KU-MEL-1 en la patogénesis de VKH.
En cuanto a los factores ambientales e infecciosos, se han correlacionado infecciones virales y enfermedades autoinmunes con la enfermedad de VKH. Virus, como Epstein-Barr y el citomegalovirus han sido propuestos como posibles factores desencadenantes de la enfermedad18. Se ha informado de VKH después de tratamiento con bacilo Calmette Guerin para melanoma, luego de cirugía de melanoma metastásico y después de heridas traumáticas en la piel2,6. Se vienen reportando casos que asocian VKH con la terapia para hepatitis C a base de interferón alfa-2b combinada o no con ribavirin21.
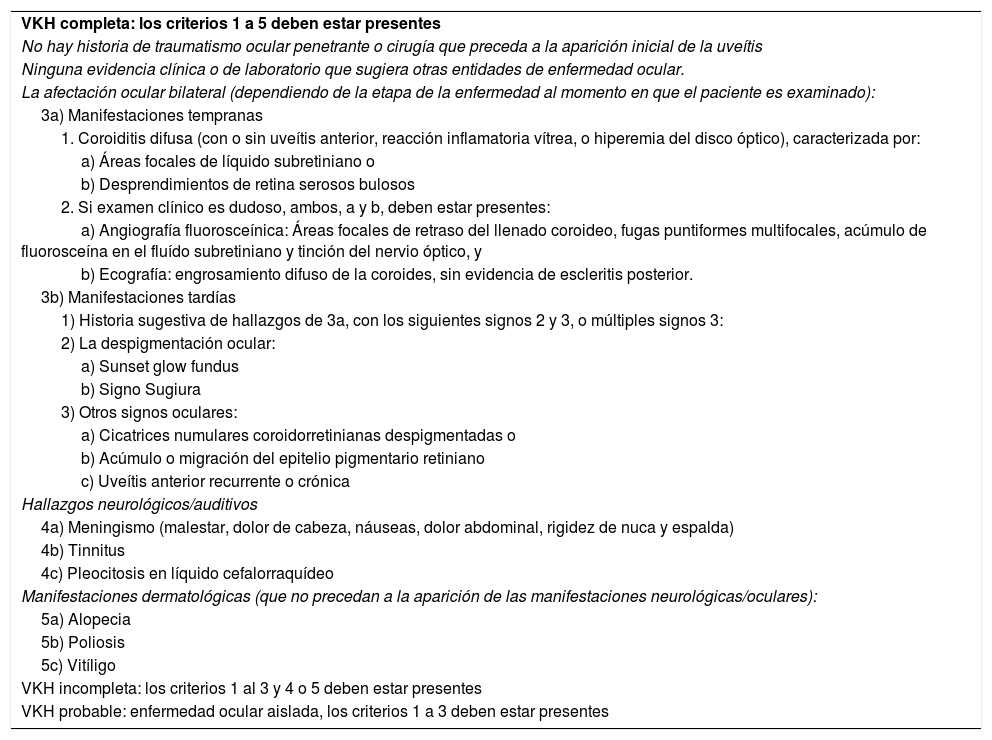
DiagnósticoLos criterios revisados de VKH han sido ampliamente aceptados desde su publicación en 20013. La tabla 2 muestra los criterios diagnósticos revisados para VKH de las etapas agudas y crónicas y dan cuenta de su naturaleza multisistémica. El diagnóstico de VKH se basa principalmente en hallazgos clínicos y se complementa con imágenes.
Criterios diagnósticos de la enfermedad de Vogt-Koyanagi-Harada
| VKH completa: los criterios 1 a 5 deben estar presentes |
| No hay historia de traumatismo ocular penetrante o cirugía que preceda a la aparición inicial de la uveítis |
| Ninguna evidencia clínica o de laboratorio que sugiera otras entidades de enfermedad ocular. |
| La afectación ocular bilateral (dependiendo de la etapa de la enfermedad al momento en que el paciente es examinado): |
| 3a) Manifestaciones tempranas |
| 1. Coroiditis difusa (con o sin uveítis anterior, reacción inflamatoria vítrea, o hiperemia del disco óptico), caracterizada por: |
| a) Áreas focales de líquido subretiniano o |
| b) Desprendimientos de retina serosos bulosos |
| 2. Si examen clínico es dudoso, ambos, a y b, deben estar presentes: |
| a) Angiografía fluorosceínica: Áreas focales de retraso del llenado coroideo, fugas puntiformes multifocales, acúmulo de fluorosceína en el fluído subretiniano y tinción del nervio óptico, y |
| b) Ecografía: engrosamiento difuso de la coroides, sin evidencia de escleritis posterior. |
| 3b) Manifestaciones tardías |
| 1) Historia sugestiva de hallazgos de 3a, con los siguientes signos 2 y 3, o múltiples signos 3: |
| 2) La despigmentación ocular: |
| a) Sunset glow fundus |
| b) Signo Sugiura |
| 3) Otros signos oculares: |
| a) Cicatrices numulares coroidorretinianas despigmentadas o |
| b) Acúmulo o migración del epitelio pigmentario retiniano |
| c) Uveítis anterior recurrente o crónica |
| Hallazgos neurológicos/auditivos |
| 4a) Meningismo (malestar, dolor de cabeza, náuseas, dolor abdominal, rigidez de nuca y espalda) |
| 4b) Tinnitus |
| 4c) Pleocitosis en líquido cefalorraquídeo |
| Manifestaciones dermatológicas (que no precedan a la aparición de las manifestaciones neurológicas/oculares): |
| 5a) Alopecia |
| 5b) Poliosis |
| 5c) Vitíligo |
| VKH incompleta: los criterios 1 al 3 y 4 o 5 deben estar presentes |
| VKH probable: enfermedad ocular aislada, los criterios 1 a 3 deben estar presentes |
Hay 3 entidades clínicas: completa, incompleta y probable, derivadas de 5 criterios. Como puede observarse en la tabla 2, la ausencia de antecedentes de trauma o cirugía ocular penetrante y de evidencia indicativa de otras enfermedades oculares es obligatoria. Además, debe documentarse la afectación ocular bilateral que indique inflamación coroidea en la fase aguda o crónica. Los hallazgos antes mencionados cumplen con el diagnóstico de probable VKH o enfermedad ocular aislada. El tipo incompleto requiere hallazgos neurológicos/auditivos o dérmicos y el tipo completo requiere ambos5. Además, los hallazgos dermatológicos no deben preceder al compromiso del sistema nervioso central ni de la enfermedad ocular.
La presentación de la enfermedad en sus formas completa, incompleta y probable muestra prevalencias variadas derivadas de diferencias étnicas, geográficas y de la duración del seguimiento. Así por ejemplo, en China se reporta prevalencia de tipo completo en el 67%, mientras que en Estados Unidos y Arabia Saudí la forma probable es más común, en cerca del 60%5.
Estudios complementariosEn la mayoría de los casos, cuando el paciente presenta manifestaciones oculares y extraoculares, el diagnóstico de la enfermedad es clínico3. Sin embargo, cuando la enfermedad se presenta con compromiso visual sin afectación extraocular, diversas pruebas complementarias han mostrado utilidad para confirmar el diagnóstico1,22. Las imágenes multimodales han proporcionado información crucial para el diagnóstico, la evaluación del tratamiento y la supervisión de las enfermedades retinocoroidales, incluyendo VKH. Las imágenes multimodales abarcan métodos no invasivos: fotografía de fondo de ojo, tomografía óptica coherente OCT, la OCT de profundidad mejorada, la biomicroscopia ultrasónica (BMU), B-scan ultrasonográfico, autofluorescencia de fondo y métodos invasivos como la angiografía fluoresceínica de fondo (AGF) y la AVI23. Su papel diagnóstico está claramente demostrado en casos típicos de VKH y es clave en la exclusión de enfermedades que pueden entrar en el diagnóstico diferencial de casos atípicos; además, las imágenes multimodales sirven como signo de valor pronóstico y tienen un papel fundamental en la monitorización de la inflamación intraocular, en la respuesta al tratamiento y en el desarrollo de complicaciones.
Los hallazgos de la AGF son muy característicos tanto en la fase aguda como en la fase crónica de la enfermedad. En la fase aguda, durante la etapa inicial de la AGF, típicamente, aparecen múltiples puntos hiperfluorescentes en el EPR, que aumentan de tamaño en la etapa tardía. Tales puntos representan acúmulo de la fluoresceína en los espacios correspondientes a desprendimientos retinianos. En la etapa tardía el angiograma muestra múltiples imágenes multimodales hiperfluorescentes. La AGF muestra en la mayoría de los pacientes hiperfluorescencia papilar y llenado coroideo anormal en parches23. La AGF en la fase crónica de VKH también es muy útil: puede mostrar alteraciones típicas en zonas de atrofia o de hiperplasia del EPR, neovascularización coroidea, anastomosis retinocoroideas y neovascularización retiniana o papilar2,23.
La AVI es un método apropiado para estudiar enfermedades coroideas, incluida la enfermedad de VKH, tanto en la fase aguda, útil en el diagnóstico temprano, como en el curso del tratamiento y la respuesta obtenida. Los hallazgos en la AVI son de utilidad para determinar si existe inflamación activa o persistente y, también, para definir si hay resolución de la enfermedad1,7.
La OCT es útil para el diagnóstico y monitorización de las imágenes multimodales exudativas maculares en la fase aguda de la enfermedad. La OCT facilita el registro de imágenes consideradas patrones clásicos de las imágenes multimodales en VKH10,13.
Mediante OCT de profundidad mejorada se ha podido medir el grosor coroideo en diferentes fases de la enfermedad y así demostrar un grosor mayor en la fase aguda y su disminución tras el tratamiento, por lo que podría ser de utilidad como marcador del grado de inflamación coroidea12,17. En pacientes con enfermedad de larga evolución (mayor a 6 meses), se observó una coroides significativamente más fina que en los controles; de hecho, este adelgazamiento coroideo, que es común en VKH, solo puede ser medido con precisión mediante OCT de profundidad mejorada: llega a determinarse que el grosor coroideo es inversamente proporcional a la duración de la enfermedad17,24.
La ecografía y biomicroscopia BMU suministran imágenes importantes para el diagnóstico de la enfermedad en sus diferentes fases, particularmente en la etapa aguda1,3. La BMU puede mostrar desprendimiento ciliocoroideo con aplanamiento de la cámara anterior en las fases iniciales de VKH6.
La RM de cerebro y de órbitas con contraste tiene utilidad cuando el paciente consulta tempranamente en el curso de la fase prodrómica o uveítica aguda. La RM de cerebro permite ver realces meníngeos y lesiones focales intraparenquimatosas. La RM de órbitas permite diferenciar la coroides y la esclera, además de identificar imágenes multimodales. En casos de VKH descarta enfermedad escleral primaria cuando se presentan dificultades diagnósticas2,25.
Los expertos consideran la PL de práctica limitada no rutinaria para el diagnóstico de VKH: es de utilidad en casos atípicos o en pacientes que consultan precozmente con pocos signos oculares y signos meníngeos positivos3,5,6.
TratamientoEl tratamiento se basa en el uso de 3 elementos: corticosteroides, terapia inmunosupresora (TIS) y modificadores de respuesta biológica.
CorticosteroidesEn VKH el tratamiento está dirigido a suprimir la inflamación coroidea aguda con el inicio temprano de corticosteroides sistémicos a dosis altas y sostenidas durante 4 a 6 meses, seguido de una disminución progresiva3.
Recientemente se ha descrito reducción del espesor de la coroides y retina después del inicio de metilprednisolona intravenosa por 3 a 5 días, en dosis que oscilan entre 200mg/día y un gramo/día, seguido de prednisolona oral 1mg/kg al día5,26. Este régimen de corticosteroides consistente en dosis alta y temprana permite una resolución de la enfermedad con menos complicaciones que con dosis más tardías y menores26. Aun así, la etapa crónica de la enfermedad puede desarrollarse hasta en un tercio de los pacientes.
También se ha usado inyección de esteroides por vía local e incluye su aplicación transeptal, inyecciones intravítreas e implantes de esteroides de liberación sostenida cuya eficacia y seguridad no se han definido con claridad27,28. Para la inflamación de la cámara anterior están indicados la prednisolona tópica al 1% y los agentes cicloplégicos, ya sea en la fase recurrente inicial o crónica, con el fin de reducir el dolor e inflamación y para prevenir las sinequias. Los corticosteroides tópicos deben ser retirados a medida que la inflamación disminuye2,5.
El empleo temprano de dosis altas de corticosteroides sistémicos también se ha mostrado eficaz para el tratamiento de las manifestaciones auditivas de VKH. De los casos de VKH con problemas auditivos, el 75% volvió a la normalidad; sin embargo, aunque muy raro, la enfermedad VKH puede causar una pérdida auditiva profunda11.
Terapia inmunosupresoraLa utilidad de la TIS está ampliamente demostrada en pacientes con problemas sistémicos autoinmunes y su uso permite reducir los efectos secundarios de la administración prolongada de corticosteroides. La Sociedad Americana de Uveítis y el panel de consenso del Grupo Internacional de Estudios sobre Uveítis aceptaron la necesidad de TIS en el VKH porque hay un resultado funcional significativamente mejor con su inicio temprano2. En un estudio de cohorte retrospectivo en Chile los autores encontraron evolución visual significativamente mejor con el inicio temprano de TIS en un subgrupo de pacientes con VKH con severo compromiso de la agudeza visual29.
La ciclosporina A, un inhibidor de la calcineurina que se dirige más específicamente a las células T, es una elección apropiada en el tratamiento de VKH. La ciclosporina ha sido el inmunosupresor más ampliamente utilizado en el cuidado de pacientes con esta enfermedad30. Con una acción similar a la ciclosporina, el tacrolimus, inhibidor de células T, es otra opción terapéutica citostática potencial. Entre los agentes antimetabolitos, el micofenolato de mofetil y la azatioprina, supresores de células T y de células B, son opciones de tratamiento confiables en pacientes VKH31. El metotrexato ha demostrado tener buen perfil de seguridad a largo plazo, con eficacia clínica en niños y adultos30. También se han reportado los efectos sinérgicos de la TIS triple agente con la combinación de prednisolona oral, azatioprina y ciclosporina en el rápido control de la inflamación y un buen resultado visual en casos severos y refractarios de VKH32.
En síntesis, la terapia TIS combinada con corticosteroides sistémicos redujo significativamente las recurrencias de uveítis, el desarrollo de complicaciones tardías y mejoró el resultado visual en comparación con otro grupo de pacientes VKH tratados con monoterapia con corticosteroides o con adición tardía de TIS.
Modificadores de respuesta biológicaAdemás de los corticoides y la TIS, las nuevas terapias modificadoras de la respuesta biológica se vienen abriendo paso en el tratamiento de la uveítis. Los anticuerpos monoclonales bloqueantes de citocinas inflamatorias específicas, como por ejemplo, el infliximab32 y adalimumab33 que se dirigen contra el factor de necrosis tumoral α (TNF-alfa) y el rituximab anti-CD2034, se vienen utilizando en uveítis refractarias a la terapia inmunomoduladora.
El factor de crecimiento del endotelio vascular (VEGF), citocina que promueve la proliferación de células endoteliales y aumenta la permeabilidad vascular, juega un papel importante en la patogénesis de las complicaciones uveíticas, como el edema macular cistoide, la neovascularización coroidea y la neovascularización de la retina35. Se ha demostrado sobreexpresión de los niveles de VEGF en el suero y en el humor acuoso de pacientes con diferentes tipos de uveítis, incluyendo VKH. La aplicación intravítrea de anticuerpos anti-VEGF, como bevacizumab y ranibizumab, inhiben la neovascularización coroidea y reducen los desprendimientos serosos de la retina en los enfermos VKH, por tanto, hoy se consideran como terapia adyuvante a la terapia sistémica convencional18.
DiscusiónVKH es una enfermedad que ha sido bien estudiada, con criterios diagnósticos precisos. A pesar de tener un patrón de presentación clínico regular y consistente, es poco conocida en Colombia. De VKH no se encontraron, durante el periodo de búsqueda, reportes de casos o revisiones en la literatura médica colombiana disponibles. Esto puede indicar que la enfermedad se presenta y es subdiagnosticada o no es reportada.
El diagnóstico clínico de VKH puede ofrecer dificultades por varias razones. En primer lugar, la enfermedad tiene 4 fases cronológicas y los pacientes al momento de la consulta inicial pueden estar cursando una fase cualquiera de la enfermedad y la demostración de la inflamación coroidomeníngea y de su caracterización en la fase correspondiente se torna un verdadero desafío. En segundo lugar, cuando la fase uveítica aguda se presenta, los pacientes alarmados por las alteraciones visuales consultan por primera vez, usualmente a oftalmología. Sin embargo, es importante tener presente que esta fase de inflamación ocular aguda frecuentemente se instaura cuando está en curso aún la fase prodrómica, con lo que se conforma un cuadro clínico caracterizado por cefalea, disacusia, tinnitus, meningismo y alteraciones oculares. Esta presentación de la enfermedad constituye un reto diagnóstico para el grupo médico. Su abordaje clínico debe incluir el estudio de la alteración oftalmológica y del compromiso neurológico y otovestibular, simultáneamente. Fue en estas condiciones de concomitancia clínica entre fase prodrómica y uveítica aguda en la que consultó la paciente del presente caso. Por tanto, en casos como este, se considera importante reconocer las diversas presentaciones de VKH enfatizando las características clínicas y paraclínicas de las 2 fases iniciales para diagnosticar la enfermedad tempranamente.
En relación con los criterios diagnósticos, oftalmólogos de áreas endémicas de VKH y expertos en uveítis de centros de referencia especializados cuestionan la necesidad de practicar PL como procedimiento diagnóstico, al considerar que el diagnóstico de la enfermedad es fundamentalmente clínico y, en casos dudosos, las imágenes de AGF y de la OCT macular y de nervio óptico ayudan a confirmar la impresión clínica y, por tanto, no conviene exponer a los pacientes a los efectos secundarios de la PL.
Contra esta opinión, los autores del presente trabajo consideran que, por un lado, el diagnóstico de la coroiditis durante la fase uveítica aguda de la enfermedad VKH no es tan evidente en un primer examen oftalmológico y, por otro, aunque en el examen oftalmológico se observara la clásica coroiditis aguda de VKH con o sin desprendimientos de retina, si el paciente tiene cefalalgia y signos de irritación meníngea el abordaje neurológico es necesario y la PL es mandatoria. De hecho, el síndrome uveomeníngeo encefálico plantea un espectro etiológico muy amplio que incluye varias enfermedades infecciosas, neoplásicas, paraneoplásicas y autoinmunes. En consecuencia, el estudio del LCR es clave para dar precisión y certeza diagnóstica. Si se demuestra pleocitosis linfocitaria en LCR, en consonancia con una meningitis aséptica, se cumple con un criterio que permite iniciar tratamiento inmunosupresor. Incluso, a un paciente con diagnóstico establecido de VKH, inmunosuprimido por el tratamiento, que en el curso de la evolución presenta cefalalgia y meningismo, se le debe practicar nueva PL para descartar neuroinfección oportunista o una meningitis aséptica recurrente. En una balance riesgo-beneficio, es mucho mayor el beneficio obtenido de la PL en comparación con sus posibles complicaciones; el temor a los efectos colaterales derivados de una PL no parece ser motivo válido para detener su realización.
ConclusionesLa revisión de tema aportó información actualizada relevante para dar respuesta a la pregunta formulada por el grupo investigador y sacar las siguientes conclusiones:
- 1.
Existen criterios diagnósticos claros y bien definidos de VKH en sus diferentes fases que permiten suministrar conocimiento suficiente para hacer el diagnóstico clínico de VKH en todo paciente de mediana edad que consulta por miodesopsias, visión borrosa, discromatopsia, cefalea, meningismo y síntomas auditivo-vestibulares. Esta es una primera forma de intervención diagnóstica válida según expertos; no obstante, el grupo investigador del presente trabajo considera que el diagnóstico clínico es admisible en zonas endémicas donde la enfermedad es frecuente, pero no en zonas donde la enfermedad es ocasional y poco conocida.
- 2.
Cuando el diagnóstico clínico de la enfermedad VKH se dificulta en casos de presentaciones atípicas o cuando el caso ocurre en áreas no endémicas, enfocar el estudio del caso a partir de un diagnóstico sindromático es una forma de intervención válida y apropiada. Es así como el diagnóstico de síndrome uveomeningoencefálico alerta al equipo de cuidado médico, implica hospitalización en centro de alta complejidad, facilita la atención simultánea de un equipo multidisciplinario y permite dar celeridad a los exámenes paraclínicos. Fue esta segunda forma de intervención la que se dio al presente caso, donde los resultados de la AGF, OCT, foto de fondo de ojo, las imágenes de RM y la PL permitieron hacer el diagnóstico de enfermedad de VKH. Para el grupo investigador esta segunda forma de intervención es pertinente y útil en regiones como la nuestra, donde la enfermedad es rara y poco conocida. En conclusión, en Colombia es necesario el uso de paraclínicos para hacer el diagnóstico de VKH.
- 3.
La revisión actualizada de la etiología y fisiopatogenia de la enfermedad VKH demuestra su naturaleza autoinmune y multisistémica, lo que obliga a un abordaje médico interespecializado. La participación de oftalmología, medicina interna, neurología, reumatología, otorrinolaringología y dermatología se hace pertinente y necesaria con fines diagnósticos y terapéuticos.
- 4.
En cuanto al tratamiento, se recomienda el uso vigoroso de corticoides en la fase uveítica aguda. La terapia basada en la evidencia recomienda el uso de pulsos de metilprednisolona de 1 gramo intravenoso al día por 3 a 5 días, seguido de prednisolona oral de 1 mg/kg al día, agregando siempre TIS tempranamente, con la ciclosporina como el fármaco más usado. Se recomienda también el uso de metotrexato, tacrolimus, azatioprina o micofenolato de mofetilo. Dosis menores de corticoides o su suspensión rápida están asociadas a complicaciones oculares severas. La aplicación intravítrea de anticuerpos anti-VEGF, como bevacizumab y ranibizumab, es recomendable porque se ha visto reducción en los desprendimientos serosos de retina en pacientes con VKH, inhibe la neovascularización coroidea y mejora el pronóstico visual. El ajuste del tratamiento con corticoide y TIS requiere del control estricto por oftalmología, reumatología o medicina interna, y no debe suspenderse antes de un año, de acuerdo con la evolución clínica y de imágenes.
Los autores declaran no tener ningún conflicto de intereses.
