



La osteomalacia oncogénica es un síndrome metabólico paraneoplásico caracterizado por hipofosfatemia debida a la pérdida renal de fosfato, con nivel bajo de vitamina D. Este trastorno está asociado con la liberación de factores fosfatúricos por células tumorales, especialmente el factor de crecimiento fibrolástico 23 (FGF23). Las neoplasias relacionadas con la osteomalacia oncogénica suelen ser tumores pequeños de linaje mesenquimatoso y pueden ser difíciles de localizar en algunos casos debido a su tamaño y ubicación poco accesible al examen físico. Presentamos a un paciente que desarrolló fracturas vertebrales y de cadera debido a osteomalacia oncogénica asociada con un tumor mesenquimatoso fosfatúrico del tejido graso profundo de la planta del pie, que finalmente se diagnosticó después de 3 años del inicio de los síntomas, cuando el tumor pudo ser localizado por el rastreo gammagráfico óseo con pentatreótido marcado con indio-111 y por las imágenes de resonancia magnética nuclear.
Oncogenic osteomalacia is a paraneoplastic metabolic syndrome characterised by a low phosphates in the blood due to renal phosphate losses with inadequately normal or low vitamin D levels. This disorder is associated with the release of tumour cell-secreted phosphaturic factor, most notably fibroblast growth factor 23 (FGF-23). The neoplasms related to oncogenic osteomalacia are usually small tumours of mesenchymal lineage, and they may be difficult to locate in the physical examination in some cases, due to their size and inaccessible location. The case is presented of a patient who developed vertebral and hip fractures due to oncogenic osteomalacia associated with a phosphaturic mesenchymal tumour of the deep fat tissue in the sole of the foot. This was finally diagnosed after 3 years of the onset of symptoms after being located by bone scintigraphy with Indium-111 labelled pentetreotide and magnetic resonance imaging.
La osteomalacia oncogénica (OO), también conocida como osteomalacia inducida por tumor, es un síndrome metabólico paraneoplásico caracterizado por hipofosfatemia debida a pérdidas renales de fosfato con nivel de 1,25-hidroxivitamina D [1,25(OH)-D] inadecuadamente normal o bajo. Se asocia con la liberación de factores previamente conocidos como «fosfatoninas» por las células tumorales, en especial el factor de crecimiento fibroblástico 23 (FGF23), que se comporta como una hormona fosfatúrica1. Las neoplasias relacionadas con este trastorno suelen ser pequeños tumores de linaje mesenquimatoso2,3. Presentamos a un paciente que desarrolló fracturas vertebrales y de cadera debido a una OO asociada a un pequeño tumor mesenquimatoso fosfatúrico del tejido adiposo profundo de la planta del pie.
Presentación del casoUn paciente varón de 48 años, originario de Perú, sin antecedentes personales ni familiares de interés, consultó a los 43 años por dolor lumbar de características mecánicas, irradiado a las regiones proximales de ambas extremidades inferiores, con debilidad muscular asociada, también de predominio proximal. Al examen físico refería dolor a la presión en los procesos espinosos lumbares L4 y L5 y en musculatura paravertebral de los mismos niveles. La fuerza muscular no fue valorable en las extremidades inferiores debido al dolor y a la dificultad para deambular; también se encontraba limitado para caminar sobre los talones y de puntillas. Los resultados de la bioquímica hemática (incluido el nivel de calcio) y el hemograma completo estaban dentro de rangos normales en este momento, por lo que no se amplió el estudio del metabolismo óseo. La resonancia magnética (RM) de la columna lumbar reveló una hernia central pequeña en L5-S1, una protrusión discal global en L4-L5 y una estenosis del canal lateral en L3-L5, por lo que los síntomas se atribuyeron a estos hallazgos.
El paciente fue tratado con una artrodesis de L3-L4-L5-S1 y descompresión por laminectomía. Después de la intervención no mostró mejoría clínica; el dolor y la debilidad en las extremidades inferiores persistieron y lo limitaban para caminar, por lo que requería usar un bastón. Fue reoperado 2 veces durante los siguientes 2 años debido a un aflojamiento del material de osteosíntesis; en la última intervención se observó «mala calidad ósea». El hueso esponjoso después del raspado era de consistencia suave, elástica y fácil de cortar con un bisturí. Los hallazgos histopatológicos de las muestras quirúrgicas fueron inespecíficos y se informaron como hueso laminar trabecular con variado tamaño de las trabéculas, con tejido adiposo intertrabecular muy escaso o reemplazado por degeneración serofibrinosa. Durante este último ingreso, los estudios radiográficos mostraron múltiples aplastamientos vertebrales lumbares y dorsales de grado ii de la clasificación de Genant y, en las caderas, secuelas de fracturas antiguas de tipo basicervical impactadas con deformidad en varo secundario y signos de coxartrosis bilateral. Ante la sospecha de que estos cambios pudiesen estar relacionados con alteraciones del metabolismo óseo, se remitió para su estudio a nuestra consulta monográfica de osteoporosis y enfermedades óseas metabólicas. Al reevaluar al paciente, negaba fracturas previas por fragilidad o antecedentes familiares de este tipo de fractura. No tenía alteraciones en la dentición, ni cambio en la coloración de la esclerótica ni hiperlaxitud ligamentosa. En este momento, al examen presentaba dolor a la presión sobre las apófisis lumbares y dorsales, así como movilidad de caderas limitada a 90° de flexión y 20° de rotación interna y externa. La analítica de sangre reveló un nivel de fosfatasa alcalina (FA) total de 342 U/l (valores normales [vn]: 53-128); FA ósea de 290 U/l (85% de FA total, vn: 32-83), nivel de calcio de 9,1mg/dl (vn: 8,7-10,3), de fósforo 1,9mg/dl (vn: 2,7-4,5), de 1,25(OH)-D<5 pg/ml (vn: 19-48), de 25(OH)-D 11 ng/ml (vn: 19,1-57,6), de parathormona 55,3 pg/ml (vn: 12-65), de osteocalcina 31,7 ng/ml (vn: 15-46) de telopéptido carboxilo terminal de colágeno de tipo i 0,824 ng/ml (vn: 0-0,550) y de propéptido N-terminal del procolágeno tipo 1 375,90 ng/ml (vn: 12-62). En la bioquímica urinaria de muestra de 24 h, el valor para la reabsorción tubular de fosfato fue de 69,91% (vn: 80-94), con aumento de fosfaturia excretada de 1.440mg/24h (vn: 400-1.300) y calciuria excretada normal (60mg/24h), sin otras alteraciones. El estudio se completó con niveles de triptasa y andrógenos, que se encontraban en rangos normales.
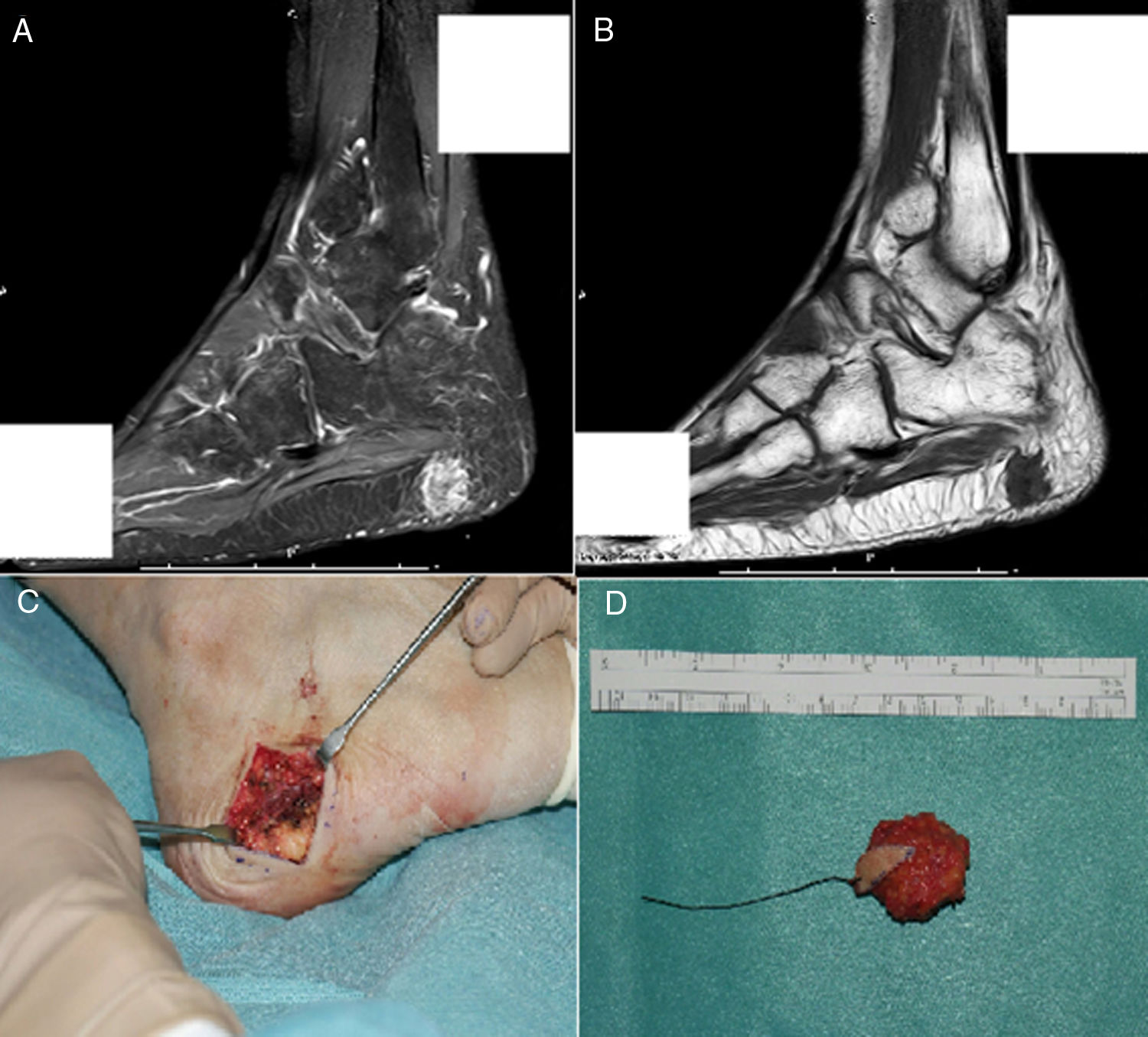
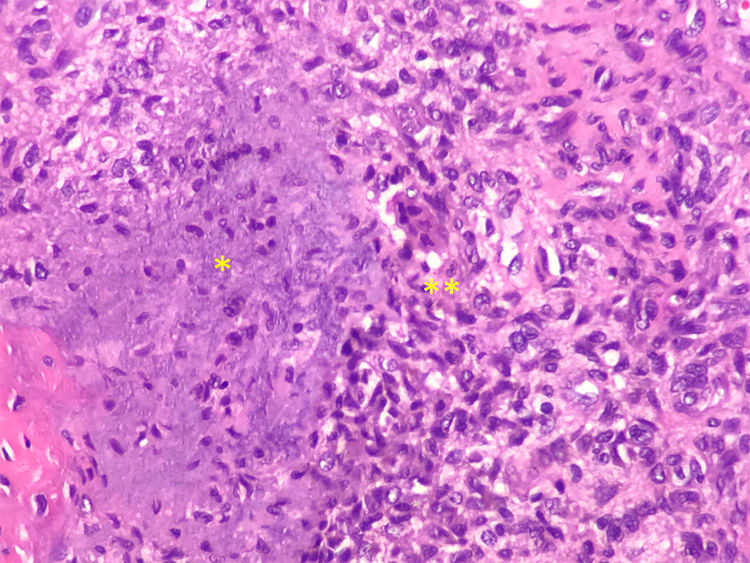
Se estableció el diagnóstico de osteomalacia hipofosfatémica y se inició el tratamiento con fosfato (4g/día) y calcitriol (0,25μg/día). Tras un mes de tratamiento, el paciente mostraba normalización del nivel de fosfato en la sangre y una leve mejoría clínica del dolor en los muslos. Dada la posibilidad de etiología tumoral, se realizó una gammagrafía ósea con pentatreótido marcado con indio-111 (111In), que mostró una captación punteada en la planta del pie derecho. Este hallazgo se confirmó por RM, y se encontró un tumor sólido de 1,7×1,4cm en la grasa plantar del retropié, con realce tras de la administración de gadolinio (figs. 1A y B). Se realizó la exéresis del tumor encapsulado con bordes libres (fig. C), que en el estudio histopatológico reveló una proliferación mesenquimatosa de crecimiento nodular, que consistía en células fusiformes, sin atipia ni pleomorfismo, bien vascularizada por vasos de tipo hemangiopericitoide, así como por células gigantes abundantes, sin atipia y con focos de calcificación basófila con un índice de proliferación muy bajo (fig. 2). Todos estos hallazgos fueron compatibles con un tumor mesenquimatoso fosfatúrico, pero el nivel de FGF23 no se pudo determinar en este caso debido a limitaciones técnicas. Después del procedimiento, el paciente presentó una normalización del nivel de fósforo en la sangre (3,1mg/dl) y pudo suspender los suplementos de fosfato en menos de un mes. Tras 3 años del inicio de los síntomas, finalmente se había podido establecer el diagnóstico de OO. En el control de las pruebas de laboratorio después de 9 meses, los niveles de fósforo (3,9mg/dl), vitamina D [25-(OH)D 27 ng/ml], reabsorción tubular de fosfato (91,4%) y fostaturia excretada (874mg/24h) se mantuvieron normales y el de FA disminuyó (132mg/dl). Se repararon las fracturas de ambas caderas mediante la colocación de prótesis totales, lo que mejoró significativamente la limitación funcional que presentaba el paciente.
 RM que muestra una lesión polilobulada, de aproximadamente 17 × 14mm, con realce tras la administración de contraste en la secuencia STIR. B) De aspecto hipointenso en T1, localizada dentro de la grasa subcutánea de la región plantar posterior, sin una clara dependencia de la fascia plantar. C) Incisión transversal en lateral plantar derecho durante el procedimiento quirúrgico. D) Aspecto macroscópico del tumor encapsulado extirpado con márgenes libres.")
A) RM que muestra una lesión polilobulada, de aproximadamente 17 × 14mm, con realce tras la administración de contraste en la secuencia STIR. B) De aspecto hipointenso en T1, localizada dentro de la grasa subcutánea de la región plantar posterior, sin una clara dependencia de la fascia plantar. C) Incisión transversal en lateral plantar derecho durante el procedimiento quirúrgico. D) Aspecto macroscópico del tumor encapsulado extirpado con márgenes libres.
 mal definida (borrosa), con presencia de células gigantes tipo osteoclasto (**). Tinción de hematoxilina y eosina (20X).")
La OO es una entidad rara, con menos de 400 casos reportados en la literatura hasta la fecha. Clínicamente, se manifiesta por dolor óseo o muscular, asociado con debilidad muscular proximal y, como en nuestro caso, fracturas por fragilidad (vertebrales, costales o de cuello femoral) que pueden aparecer en más del 80% de los pacientes antes del diagnóstico2. Debido a la naturaleza no específica de los síntomas, la duración media hasta el diagnóstico de la OO es de alrededor de 5 años, dependiendo de la serie, y puede haber un retraso incluso hasta de 20 años2,3.
La sospecha diagnóstica de OO se establece en función de los hallazgos de laboratorio característicos: hipofosfatemia con aumento de la excreción de fosfato urinario, aumento del nivel de FA y valor deficitario o normal de 1,25(OH)-D, en presencia de calcemia normal. La parathormona puede ser normal o estar elevada en el contexto de hiperparatiroidismo secundario a la deficiencia de vitamina D1. Todos estos hallazgos están relacionados con la producción de FGF23 de las células tumorales, que actúa sobre todo en los túbulos renales y aumenta la excreción renal de fosfato, además de disminuir la producción de 1,25(OH)-D4. Sin embargo, en vista de estos hallazgos analíticos, se debe hacer un diagnóstico diferencial con otras causas de osteomalacia hipofosfatémica hereditaria o adquirida5, incluidas toxinas (aluminio, arsénico), fármacos (hierro intravenoso, antirretrovirales análogos de nucleótidos) y el síndrome de Fanconi1,6,7, que fueron excluidos en este paciente tras el estudio de aminoaciduria, glucosuria y gases en sangre arterial, todos ellos normales. Otros trastornos hereditarios, originados por un exceso de FGF23, como la osteomalacia hipofosfatémica autosómica dominante, la relacionada con el cromosoma X o la autosómica recesiva, producen hallazgos bioquímicos indistinguibles de la OO4, pero el inicio de la enfermedad en la adultez, los síntomas de debilidad muscular proximal y el dolor en miembros inferiores, así como la ausencia de antecedentes familiares o de trastornos metabólicos previos descartaron esta posibilidad en nuestro caso.
La mayoría de los tumores asociados con OO son neoplasias benignas de origen mesenquimatoso y crecimiento lento, generalmente únicas y localizadas en huesos (alrededor del 30%) o en tejidos blandos (alrededor del 60%)2,3. Se distribuyen sobre todo en miembros inferiores (55-60%) y con menos frecuencia en cabeza y cuello (23-30%) y miembros superiores (alrededor de 5%)2,3. Los tumores mesenquimales reportados en la literatura incluyen el hemangiopericitoma, el tumor óseo de células gigantes, el hemangioma esclerosante, el angiolipoma y el grupo de tumores clasificados en la actualidad como tumores mesenquimatosos fosfatúricos, con varios subtipos diferentes: tumores similares a osteoblastomas, tumores similares a fibromas osificantes, tumores similares a los fibromas no osificantes y el subtipo PMTMCT (tumor fosfatúrico mesenquimatoso con tejido conjuntivo mixto), que es el tipo más frecuente y comprende aproximadamente el 70-80% de los tumores mesenquimales relacionados con la OO.
Desde un punto de vista histológico, muestran un aspecto polimorfo, compuesto por una mezcla de células fusiformes, células gigantes de tipo osteoclasto, vasos sanguíneos prominentes, matriz cartilaginosa y hueso metaplásico8,9. Por otro lado, se han descrito variantes malignas, cuya histología muestra un aspecto más sarcomatoide (mayor atipia y actividad mitótica). Las metástasis se han descrito en menos del 5% en estos casos y son, en general, pulmonares1,8,10. Debido a que suelen ser tumores pequeños y a menudo no presentan síntomas locales ni son evidentes en el examen físico, las técnicas de diagnóstico funcional son de especial interés. Es importante un rastreo del cuerpo completo, ya que es posible que se encuentren en una parte distal de las extremidades11.
Es común que estos tumores expresen receptores de somatostatina, por lo que se ha utilizado la gammagrafía con un análogo de esta hormona (pentatreótido) marcado con 111In para su detección12. Otras técnicas utilizadas son la tomografía por emisión de positrones (PET/CT) con fluoro-desoxiglucosa y, más recientemente, la PET/CT con Ga68-DOTA-1-NaI3-octreótida (DOTANOC). También se ha utilizado el muestreo venoso de FGF23, para lo cual se realizan determinaciones venosas en las 4 extremidades y se determina el nivel de FGF23 en cada una de ellas, para facilitar la localización del tumor13,14.
La identificación de áreas hipermetabólicas sospechosas requiere confirmación anatómica con MRI o tomografía computarizada, ya que en algunos casos la captación puede estar relacionada con fracturas. Si hay un cuadro clínico compatible, pero el tumor no ha sido localizado, se recomienda repetir las pruebas en 1-2 años2,3.
El tratamiento de elección en estos casos es la resección quirúrgica completa del tumor, que es curativa, ya que de lo contrario pueden reaparecer localmente15. Después de la intervención, la recuperación del nivel de fosfatemia se observa generalmente entre los 5 y 10 días y el de FA entre los 9 y 24 meses2. Si la normalización de la fosfatemia no se produce o disminuye nuevamente, debe considerarse una recurrencia local o metástasis15. Cuando no se consigue localizar el tumor o no es resecable, el tratamiento médico con suplementos de fósforo debe iniciarse en una dosis de 1-3g/día, dividida en 4-6 dosis, y calcitriol 15-60 ng/kg /día, monitorizando la respuesta al tratamiento, ya que es posible la aparición de un hiperparatiroidismo secundario1. Entre los tratamientos alternativos a la cirugía, en el caso de tumores no resecables, se ha utilizado la ablación por radiofrecuencia16. También se han notificado casos tratados con octreótido subcutáneo17 y se está evaluando el uso de anticuerpos monoclonales contra el FGF23 (KRN23), que han demostrado eficacia en pacientes con osteomalacia relacionada con el cromosoma X18.
ConclusionesLa OO es un desafío diagnóstico importante, ya que suele seguir un curso insidioso y sus síntomas no son específicos. Aunque generalmente es causada por tumores mesenquimales benignos, es importante considerarla en presencia de alteraciones bioquímicas compatibles, ya que puede ser una causa significativa de morbilidad, sobre todo en pacientes jóvenes, debido al desarrollo de fracturas por insuficiencia y la consiguiente discapacidad asociada. Aunque la determinación de los niveles de FGF23 orienta mucho el diagnóstico de OO, prima la clínica del paciente y todos los pasos en la búsqueda del diagnóstico, incluyendo los datos de laboratorio y pruebas de imagen, tanto funcionales, como la gammagrafía ósea, como morfológicas, como la RM, que permiten identificar y localizar los tumores asociados con OO de una manera precisa, ya que no suelen ser accesibles al examen físico debido a sus pequeñas dimensiones y a ubicarse en planos profundos, especialmente en miembros inferiores.
Conflicto de interesesLos autores declaran no tener conflictos de intereses.
