



La enfermedad de Erdheim-Chester (EEC) es una histiocitosis de células no Langerhans infrecuente. Se caracteriza por la infiltración de varios órganos y tejidos por histiocitos espumosos con un curso clínico heterogéneo que varía desde formas leves hasta aquellas diseminadas con un comportamiento progresivo y letal. Se presenta el caso de una paciente que debutó con un síndrome cerebeloso asociado con patología autoinmune. En el curso evolutivo, la refractariedad a glucocorticoides y la manifestación clínica con afectación ósea en forma de osteoesclerosis simétrica de huesos largos fueron determinantes para la sospecha de esta entidad. Revisamos artículos científicos con el metabuscador PubMed, empleando las palabras clave «erdheim chester disease», «erdheim chester and nervous system» y «autoimmunity and erdheim chester disease», y seleccionamos trabajos con un mayor énfasis en la presentación clínica con afectación neurológica y patología autoinmune asociada. Los avances en la patogénesis de la EEC han permitido conocer la naturaleza de la enfermedad, así como la utilización de terapias dirigidas. Interesa tener presente esta entidad, así como las patologías a las que se asocia con frecuencia, con el objetivo de obtener un diagnóstico precoz y un mejor abordaje clínico.
Erdheim Chester disease (ECD) is a rare non-Langerhans cell histiocytosis. It is characterized by the infiltration of various organs and tissues by foamy histiocytes with a heterogeneous clinical course that varies from mild forms to disseminated forms with progressive and lethal behaviour. The case of a patient who presented with a cerebellar syndrome associated with autoimmune pathology is presented. In the course of the disease, refractoriness to glucocorticoids and clinical manifestation with bone involvement in the form of symmetrical osteosclerosis of long bones were determining factors for suspicion of this entity. We reviewed scientific articles through the PubMed metasearch engine with the keywords «erdheim chester disease», «erdheim chester and nervous system», and «autoimmunity and erdheim chester disease», selecting those with greater emphasis on clinical presentation with neurological involvement and associated autoimmune pathology. Advances in the pathogenesis of ECD have allowed us to understand the nature of the disease, as well as the use of targeted therapies. It is interesting to keep this entity in mind, as well as the pathologies with which it is frequently associated, with the objective of an early diagnosis and a better clinical approach.
Las histiocitosis son trastornos raros, caracterizados por la acumulación de macrófagos, células dendríticas o células derivadas de monocitos en diversos tejidos y órganos tanto de adultos como en edad pediátrica. La clasificación actual divide estos trastornos en cinco grupos, basados en criterios clínicos, radiológicos, patológicos, genéticos o las características moleculares.
La enfermedad de Erdheim-Chester (EEC) pertenece al grupo «L» (Langerhans), junto con la histiocitosis de células de Langerhans (HCL) y el xantogranuloma juvenil extracutáneo. Se diferencia de estas entidades por la presentación clínica apoyada en la histología y fundamentalmente por las características inmunohistoquímicas y de análisis molecular de los histiocitos. Así, se considera un trastorno hematopoyético clonal infrecuente que se caracteriza por la infiltración multiorgánica de histiocitos espumosos, inflamación crónica y fibrosis.
Se presenta el caso de una paciente que debutó con un cuadro neurológico asociado con una patología autoinmune. Tras la revisión de la literatura científica disponible, se objetiva que, si bien la patogénesis de esta entidad no se conoce por completo, el descubrimiento de las mutaciones clonales que activan las vías de la proteína cinasa activada por mitógenos (MAPK) no solo ha permitido esclarecer la naturaleza de esta patología, sino que además tiene implicaciones terapéuticas. Por otro lado, se evidencia una alta prevalencia de autoinmunidad clínica o biológica asociada, que aun cuando puede comportar complejidad al diagnóstico, supone un nuevo enfoque de cara a esclarecer los mecanismos etiopatogénicos de esta entidad.
Observación clínicaMujer de 55 años con antecedente de probable enfermedad indiferenciada del tejido conectivo por clínica de xeroftalmia, aftas orales, Raynaud y anticuerpos antinucleares (ANA) positivos 1/640. Presenta inestabilidad de la marcha y disfagia de un año de evolución; en la exploración física manifiesta disartria moderada, hiperreflexia y ataxia apendicular y de la marcha.
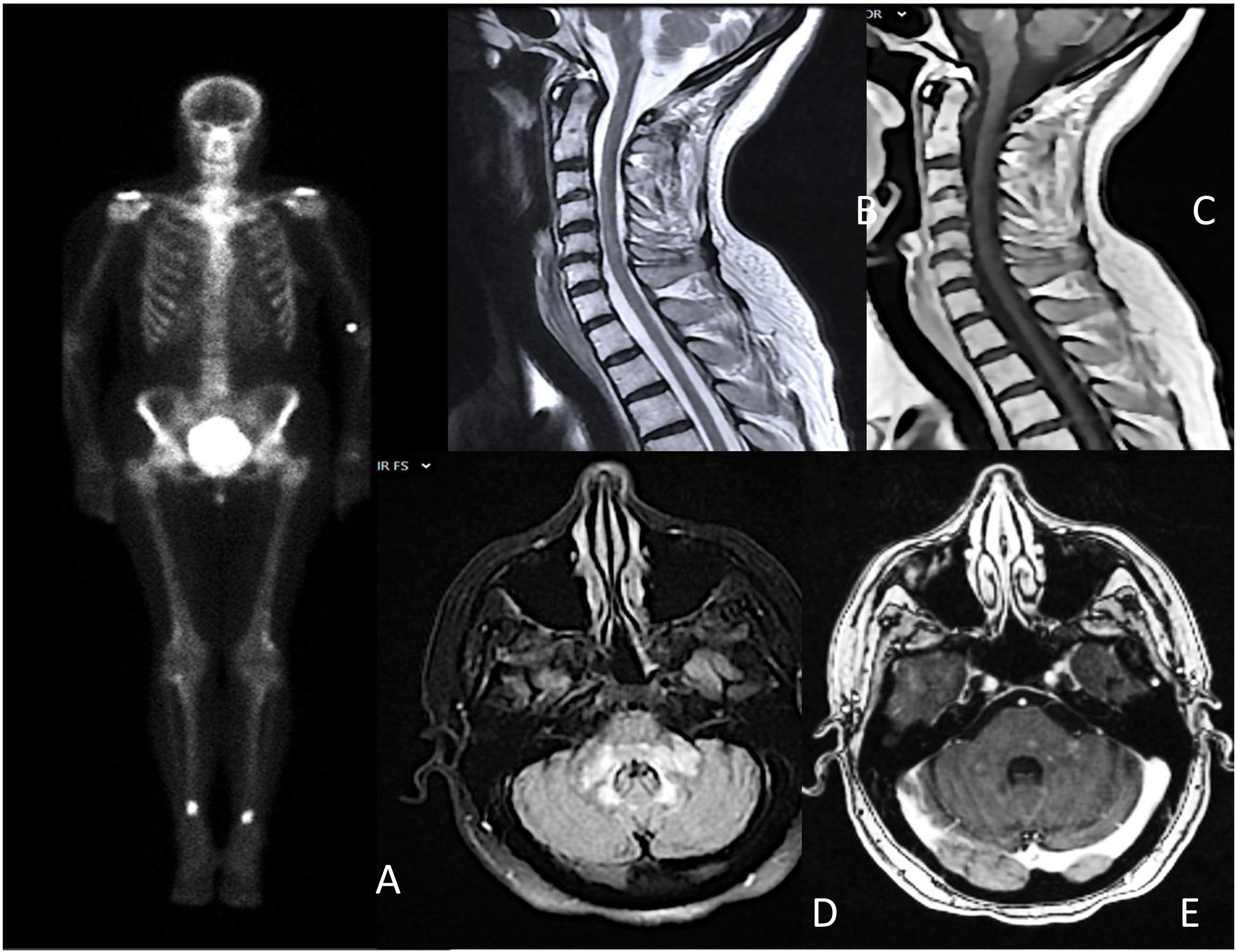
Se realizó el test de Schirmer y una biopsia de glándula salival menor, cumpliendo con los criterios de la Liga Europea Contra el Reumatismo (EULAR) para el síndrome de Sjögren (SS). En la resonancia magnética cerebral destacaron áreas hiperintensas con un discreto realce de contraste en el tronco cerebral y mielitis longitudinal extensa a nivel cervical (fig. 1). El resto de los estudios, incluido el análisis de líquido cefalorraquídeo y la tomografía de cuerpo entero fueron normales. Ante la sospecha de un proceso inflamatorio relacionado con el SS, se iniciaron glucocorticoides a dosis altas.
 Gammagrafía ósea que muestra depósitos hipercaptantes simétricos en diáfisis tibial y femoral distal; B y C) mielitis transversa longitudinal extensa C3-C6, sin captación de contraste; D y E) hiperintensidad parcheada y difusa en bulbo, protuberancia y pedúnculos cerebelosos con áreas de captación intraparenquimatosa parcheada.")
A) Gammagrafía ósea que muestra depósitos hipercaptantes simétricos en diáfisis tibial y femoral distal; B y C) mielitis transversa longitudinal extensa C3-C6, sin captación de contraste; D y E) hiperintensidad parcheada y difusa en bulbo, protuberancia y pedúnculos cerebelosos con áreas de captación intraparenquimatosa parcheada.
Durante la evolución, la paciente presentó un empeoramiento clínico-radiológico asociado con un cuadro de diabetes insípida. Se completó el estudio con una gammagrafía ósea en la que se observó hipercaptación simétrica en diáfisis femoral y diafiso-metafisiaria tibial distal. En la biopsia de las lesiones se identificó la presencia de histiocitos espumosos con inmunotinción positiva CD68, compatible con una EEC. En el estudio molecular se encontró negatividad para la mutación V600E.
Tras iniciar un tratamiento con interferón alfa (IFN-α) pegilado, la paciente presentó estabilidad clínica. Durante la evolución desarrolló un cuadro de otoxicidad secundaria al interferón y requirió por ello un cambio de tratamiento por cladribina. En la actualidad, mantiene una situación de estabilidad en seguimiento clínico y radiológico periódico.
DiscusiónLa EEC es una patología poco común de la que se han descrito 1.500 casos en la literatura desde su descripción en 1930, aunque su prevalencia y su incidencia exacta no se conocen con certeza. Afecta predominantemente a adultos con una edad media al diagnóstico de 55 años y una ratio hombre/mujer de 1,51.
El espectro clínico es heterogéneo y el curso varía según los órganos afectados, desde formas poco sintomáticas a formas diseminadas y letales. La afectación cardiovascular y del sistema nervioso central (SNC) conlleva un peor pronóstico2,3.
La manifestación más frecuente, en más del 95% de los pacientes, es la osteoesclerosis de la diáfisis y la metáfisis de huesos largos en las extremidades inferiores. La diabetes insípida, presente hasta en un tercio de los casos4,5, a menudo es la primera manifestación de la enfermedad. La afectación neurológica, en más de la mitad de los pacientes6, incluye la afectación intraparenquimatosa con predominio en la fosa posterior7, afectación vascular y meníngea, siendo infrecuente la infiltración intramedular. Destaca también la afectación pericárdica, la infiltración circunferencial de la aorta, de la grasa perirrenal y la fibrosis retroperitoneal.
El diagnóstico de EEC se establece con histología y fenotipo de histiocitos en el contexto clínico y radiológico apropiado. La rentabilidad de la biopsia varía según el tejido estudiado y son frecuentes varios intentos para conseguir la confirmación diagnóstica. Este, entre otros factores, condiciona el retraso diagnóstico, cuya duración media es de uno a 2,7 años1,4.
La biopsia es imprescindible pero la histología no es específica de la enfermedad, se requieren técnicas de inmunohistoquímica y de análisis molecular para el diagnóstico. La histopatología se caracteriza por la presencia de histiocitos mononucleados espumosos con núcleo pequeño, histiocitos multinucleados o células de Touton, fibrosis, además de la presencia de linfocitos, células plasmáticas y neutrófilos reactivos. En la EEC, los histiocitos presentan inmunotinción positiva para CD68 y CD163 y son negativos para CD1a8.
La detección de mutaciones que involucran genes de la vía de las cinasas RAS-RAF-MEK-ERK (vía MAPK), tanto en la HCL como en la EEC, supuso la redefinición de estas entidades, hoy identificadas como neoplasias proliferativas. Destaca en más de la mitad de los casos de EEC, la presencia de la mutación V600E del protooncogén B-fibrosarcoma rápidamente acelerado (BRAF). La búsqueda de estas mutaciones en la práctica clínica es útil y se recomienda para confirmar diagnósticos difíciles y en pacientes con fracaso de tratamientos de primera línea8.
La EEC y la HCL, ambas pertenecientes al grupo «L» de la clasificación actual de histiocitosis, tienen diferencias no solo en cuanto a la presentación clínica: la HCL tiene una mayor incidencia en edad pediátrica y, desde el punto de vista histológico, se caracteriza por histiocitos patológicos mononucleados con núcleos en forma de grano de café o de riñón, determinación mediante microscopia electrónica de gránulos de Birbeck citoplasmáticos, así como inmunotinción positiva para CD1a y CD207.
A pesar de estos hallazgos, la patogénesis de la EEC no está clara. Se ha demostrado la implicación de una vía proinflamatoria de citocinas como posible responsable del reclutamiento y la activación de histiocitos patológicos1,9. Se han visto implicados niveles elevados de IFN-α, un regulador de la inflamación, así como de interleucina (IL) 12, proteína quimiotáctica de monocitos-1 y niveles reducidos de IL-4 e IL-7, lo que sugiere una disfunción inmunitaria sistémica subyacente que involucra a las células T auxiliares6.
No está claro si las modificaciones de estas citocinas actúan como una causa o una consecuencia de los cambios patológicos de los histiocitos, aun cuando el microambiente alrededor de estos, con el reclutamiento de células inmunes, puede participar en la inducción de la autoinmunidad10. De esta forma, la activación inmunitaria sistémica y local evidenciada no solo se propone como impulsora del daño orgánico, sino que puede explicar la alta prevalencia de autoinmunidad asociada, tanto clínica como biológica, presente hasta en el 12% de los casos en series de pacientes, entre las que destacan la tiroiditis autoinmune, el SS y el lupus eritematoso sistémico5,11.
La mayoría de los pacientes con EEC requieren tratamiento, excepto aquellos con enfermedad asintomática o levemente sintomática. Se ha descrito la administración de varios regímenes terapéuticos que implicaban quimioterapias citotóxicas o inmunosupresoras, incluidos los alcaloides de la vinca, antraciclinas, ciclofosfamida, micofenolato de mofetilo y quimioterapia en dosis altas con trasplante autólogo de células madre, pero su eficacia clínica es limitada. Por otro lado, los corticosteroides pueden reducir el edema de forma aguda, pero no se consideran una monoterapia eficaz.
Las opciones de tratamiento se determinan en función de las características clínicas y el estado mutacional. En los tratamientos de primera línea se incluyen regímenes basados en el IFN-α, que han demostrado una mejoría en la supervivencia, aunque esta es variable según la afectación orgánica; además, pueden presentarse efectos adversos como depresión y fatiga, siendo mejor toleradas las formas pegiladas. Por otro lado, la anakinra, un antagonista de IL-1 recombinante, y la cladribina han mostrado eficacia moderada en series de pacientes con respuesta clínica y radiológica5,11.
El descubrimiento de la implicación de la vía MAPK ha permitido el abordaje con terapias dirigidas como los inhibidores de BRAF (vemurafenib y dabrafenib) e inhibidores de MEK (cobimetinib y trametinib), generalmente en casos con manifestaciones graves de la enfermedad o tras el fracaso de los fármacos convencionales2,8. Su utilización en series de pacientes ha demostrado que son eficaces, sin embargo, su toxicidad es considerable y remarca la necesidad de una monitorización estrecha de los efectos secundarios relacionados con el tratamiento. El vemurafenib puede causar toxicidad cutánea, anomalías de la conducción cardiaca, artralgias y efectos secundarios gastrointestinales, mientras que el cobimetinib se relaciona con rabdomiólisis, retinopatía y erupción acneiforme.
Las terapias dirigidas han supuesto una alternativa prometedora en el tratamiento de la EEC, no obstante, los resultados exigen un uso cuidadoso de estos medicamentos, en un entorno clínico adecuado y dejan entrever que aún quedan cuestiones por resolver; se necesitan más estudios para evaluar mejor la eficacia de la combinación de terapias, así como estudiar la eficacia de nuevos fármacos con mejor difusión en el SNC u otros mecanismos de acción12.
ConclusiónLa EEC se debe tener presente como posibilidad diagnóstica en cuadros neurológicos con neuroimagen de carácter inflamatoria refractaria al tratamiento inmunomodulador. La patología autoinmune se asocia con frecuencia y su manifestación puede solaparse con el cuadro clínico de la EEC. Los avances en el conocimiento de la etiopatogenia resultan prometedores y repercuten tanto en el diagnóstico como en el tratamiento de esta patología, que, si bien se mantiene infrecuente, se conoce cada vez más con un número mayor de diagnósticos y expectativas para los pacientes que la padecen.
Consideraciones éticasSe contó con el consentimiento verbal y escrito por parte de la paciente para la utilización de datos clínicos, así como de las pruebas complementarias realizadas, para esta finalidad.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
