


El síndrome de Kabuki (SK) es una rara enfermedad genética que cursa con importantes alteraciones cardiacas e inmunológicas. La mayoría de los pacientes son diagnosticados en los primeros años de vida pese a que la edad de inicio no está bien definida. Los pacientes afectos presentan infecciones de repetición debido a su inmunosupresión pudiendo llegar a un estado de agranulocitosis. Aunque su pronóstico es bastante favorable, su esperanza de vida viene condicionada por este tipo de complicaciones. Por ello, conocer sus características analíticas particulares resulta interesante desde el punto de vista del laboratorio de hematología para contribuir en su orientación diagnóstica y en el seguimiento de los mismos. Presentamos el caso de un paciente diagnosticado de SK.
Kabuki syndrome (KS) is a rare genetic disease that usually involves significant cardiac and immunological disorders. Most patients are diagnosed in the first years of life, despite the fact that the age of onset is not well-defined. Affected patients have recurrent infections due to their immunosuppression, and may reach a state of agranulocytosis. Although their prognosis is quite favourable, their life expectancy is determined by these types of complications. Therefore, to understand its particular analytical characteristics is interesting from the point of view of the Haematology Laboratory to contribute to their diagnosis and follow-up. The case is presented of a patient diagnosed with KS.
El Kabuki es una forma de teatro japonés tradicional que se caracteriza por su drama estilizado y el uso de maquillajes elaborados en los actores. A modo de alegoría, en 1981 Niikawa et al.1 y Kuroki et al.2 describieron en Japón de manera simultánea un raro desorden congénito de causas desconocidas asociado a múltiples anomalías congénitas y retraso mental bautizándolo como el síndrome de Kabuki (SK)3 al recordar las caras de los pacientes a las antiguas máscaras japonesas usadas en el teatro Kabuki.
Desde entonces hasta nuestros días, el SK ha sido observado en todos los grupos étnicos con una prevalencia que oscila entre 1:32.000 y 1:60.0004. No obstante, son menos de 400 los casos publicados en todo el mundo por lo que se considera una entidad infradiagnosticada5. Aunque la incidencia global de la enfermedad es desconocida, en España se estima una media de 1.500 afectados. Por este motivo, en el año 2014 se constituyó la Asociación Síndrome de Kabuki con el objetivo de dar apoyo y cuidados a las personas afectas de esta enfermedad bajo el lema «Los niños con Síndrome de Kabuki no son invisibles, pero tú puedes hacerlos más visibles. Haz lo posible, hazlo posible»6.
Descripción del casoPaciente de 6 años que acude a urgencias por otitis serosas de repetición.
Como antecedentes personales destaca un diagnóstico prenatal de hipoplasia pulmonar derecha con drenaje venoso anómalo parcial, sin cardiopatía estructural, asociado a un triple screening patológico con un cariotipo 46XX. Anteriormente la paciente presentó un episodio de fiebre a los 3 meses de edad acompañado de síntomas catarrales y gingivoestomatitis que cedió con tratamiento con antibiótico (azitromicina) durante 3 días y otro episodio febril a los 7 meses de edad asociado a un absceso cutáneo en glúteo izquierdo donde se aisló en cultivo puro Pseudomonas aeruginosa. Este último episodio también fue resuelto con tratamiento antibiótico (piperacilina-tazobactam) durante 7 días.
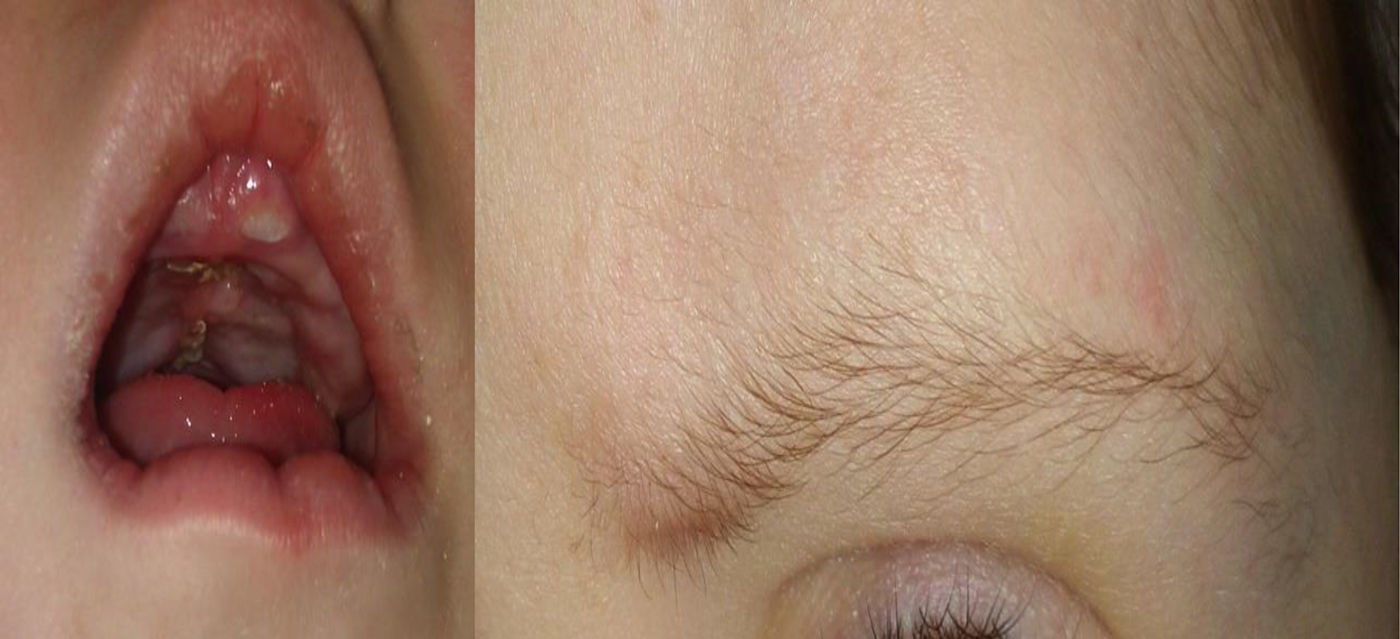
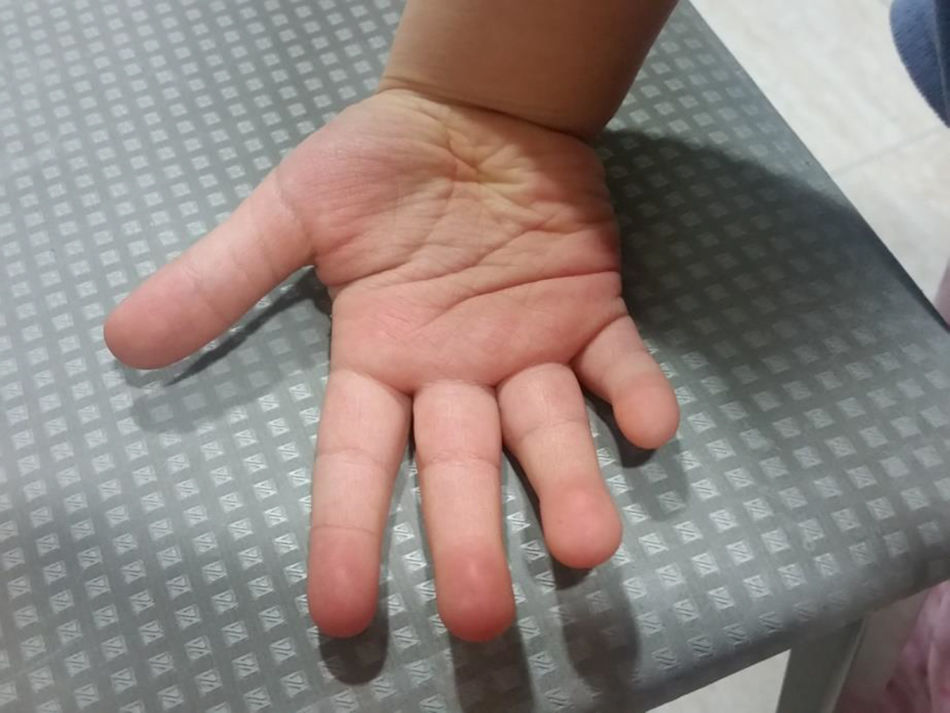
En la exploración se objetiva un retraso del crecimiento y retraso madurativo global con un fenotipo peculiar: hipotonía axial, cara tosca con cejas escasamente pobladas en el tercio distal (fig. 1), columnela corta, orejas grandes, fisura palatina (reconstrucción a los 13 meses) (fig. 1), sinfalangismo distal, filtro corto, labio superior arqueado (fig. 1) y persistencia de almohadillas en el pulpejo de los dedos (fig. 2).
. Fisura palatina (reconstruida) y labio superior arqueado de la paciente (izquierda). Rasgos fenotípicos asociados al síndrome de Kabuki.")

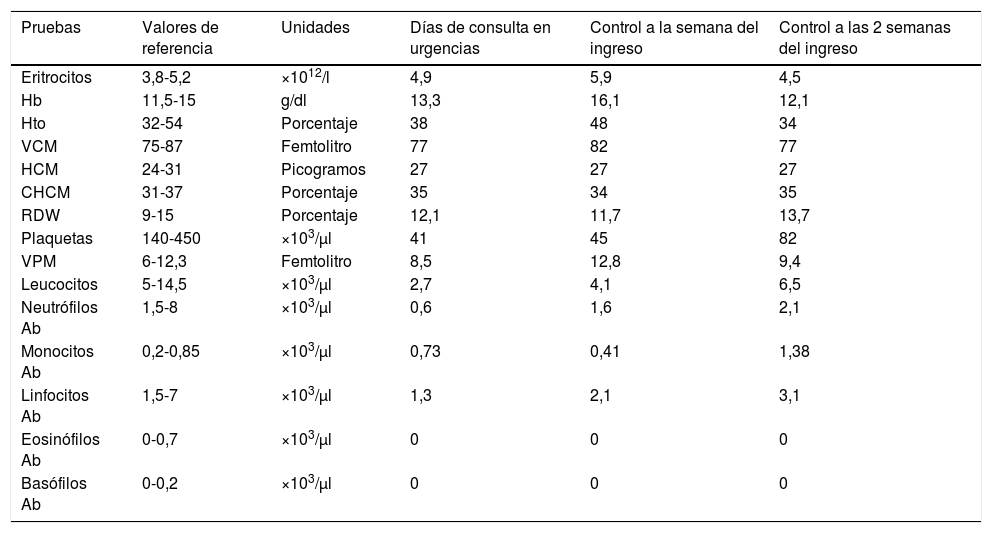
En las pruebas complementarias destaca una agranulocitosis asociada a trombocitopenia (tabla 1) que precisó ingreso hospitalario y tratamiento con factor de estimulante de colonias de granulocitos (dosis inicial de 35μg en las primeras 24h; 20μg cada 24h durante los 4 días siguientes) por el acusado descenso de los niveles plaquetarios (tabla 1).
Hematimetría de la paciente en el momento de la llegada a urgencias y su evolución
| Pruebas | Valores de referencia | Unidades | Días de consulta en urgencias | Control a la semana del ingreso | Control a las 2 semanas del ingreso |
|---|---|---|---|---|---|
| Eritrocitos | 3,8-5,2 | ×1012/l | 4,9 | 5,9 | 4,5 |
| Hb | 11,5-15 | g/dl | 13,3 | 16,1 | 12,1 |
| Hto | 32-54 | Porcentaje | 38 | 48 | 34 |
| VCM | 75-87 | Femtolitro | 77 | 82 | 77 |
| HCM | 24-31 | Picogramos | 27 | 27 | 27 |
| CHCM | 31-37 | Porcentaje | 35 | 34 | 35 |
| RDW | 9-15 | Porcentaje | 12,1 | 11,7 | 13,7 |
| Plaquetas | 140-450 | ×103/μl | 41 | 45 | 82 |
| VPM | 6-12,3 | Femtolitro | 8,5 | 12,8 | 9,4 |
| Leucocitos | 5-14,5 | ×103/μl | 2,7 | 4,1 | 6,5 |
| Neutrófilos Ab | 1,5-8 | ×103/μl | 0,6 | 1,6 | 2,1 |
| Monocitos Ab | 0,2-0,85 | ×103/μl | 0,73 | 0,41 | 1,38 |
| Linfocitos Ab | 1,5-7 | ×103/μl | 1,3 | 2,1 | 3,1 |
| Eosinófilos Ab | 0-0,7 | ×103/μl | 0 | 0 | 0 |
| Basófilos Ab | 0-0,2 | ×103/μl | 0 | 0 | 0 |
Ab: absolutos; CHCM: concentración de hemoglobina corpuscular media; dl: decilitro; g: gramos; Hb: hemoglobina; HCM: hemoglobina corpuscular media; Hto: hematocrito; mm3: milímetro cúbico; RDW: ancho de distribución eritrocitaria; VCM: volumen corpuscular medio; VPM: volumen plaquetario medio; μl: microlitro.
Ante el fenotipo peculiar de la paciente y la inmunodeficiencia asociada, se sospecha de un SK, por lo que se realiza estudio genético.
Tras la obtención del ADN de la paciente se amplificaron mediante PCR todos los exones codificantes y zonas intrónicas adyacentes del gen KMT2D (MLL2) analizando ambas cadenas de los fragmentos amplificados mediante secuenciación tipo Sanger (Applied Biosystems® 3500DX Genetics Analyzer).
La paciente presentó en heterocigosis la mutación c.7650delT (p.V2551SfsX32) en el exón 31 del gen KMT2D (MLL2). Esta mutación está asociada al SK y supone un cambio de marco de lectura a partir del codón 2551; lo que provoca la aparición de un codón de terminación prematuro y la producción de una proteína más corta de lo normal (2.581 aminoácidos de la proteína mutada frente a los 5.537 aminoácidos de la proteína normal)7.
Tras el estudio genético de los progenitores y ante la ausencia de mutación en los mismos se diagnostica la aparición de la mutación como de novo. Puesto que el modo de herencia del SK descrito para la mutación del gen KMT2D (MLL2) es autosómica dominante y su penetrancia parece ser completa, la presencia de esta mutación en heterocigosis se considera diagnóstica para el desarrollo de esta enfermedad.
Discusión y conclusionesEl SK ocurre de forma esporádica en la mayoría de casos. En un 45-80% de los casos se encuentra mutado el gen KMT2D (MLL2) con un patrón de herencia autosómico dominante. Este gen proporciona instrucciones para la fabricación de una enzima llamada lisina específica metiltransferasa 2D (LEMT2D) que se encuentra en muchos órganos y tejidos del cuerpo. La LEMT2D funciona como una histona metiltransferasa; enzima que modifica a las proteínas histonas mediante la adición de un grupo metilo controlando de esta manera la expresión de diferentes genes que parecen estar involucrados en el proceso de desarrollo8.
En una minoría de pacientes (entre un 2 y un 6% los casos de SK), la mutación se encuentra en el gen KDM6A de herencia ligada a X. Este gen proporciona instrucciones para la fabricación de una enzima llamada lisina específica desmetilasa 6A (LED6A); enzima encargada de eliminar los grupos metilo de ciertas histonas. Como la LEMT2D, la LED6A regula la actividad de ciertos genes, y la investigación sugiere que las 2 enzimas trabajan juntas para controlar ciertos procesos de desarrollo madurativo.
Las mutaciones del gen KMT2D y KDM6A asociadas con el SK conducen a la ausencia de la enzima funcional correspondiente. La falta de enzimas producidas a partir de estos genes altera la metilación normal de las histonas desregulando la expresión génica en los distintos órganos y tejidos del cuerpo, traduciéndose de esta manera en las anomalías en el desarrollo y función características del SK.
En el resto de casos con SK no se han identificado mutaciones en los genes KMT2D o KDM6A siendo la causa del trastorno en estos individuos aún desconocida.
Actualmente no se han establecido criterios para el diagnóstico clínico. El diagnóstico se basa en la observación clínica de 5 hallazgos cardinales que son9:
- 1.
Hallazgos craneofaciales
- 2.
Retraso en el crecimiento posnatal
- 3.
Anomalías esqueléticas
- 4.
Persistencia del almohadillado fetal
- 5.
Déficit intelectual
El análisis molecular por su parte confirma el diagnóstico clínico haciendo posible el diagnóstico prenatal cuando se conoce la mutación responsable9.
El SK presenta un espectro clínico amplio y variable. Los rasgos craneofaciales incluyen: fisuras palpebrales alargadas con eversión del tercio externo del párpado inferior; cejas arqueadas y espesas, pero en su tercio lateral escasamente pobladas o con zonas vacías en forma de pequeñas muescas como presentaba la paciente (fig. 1); columnela corta con la punta nasal plana; orejas grandes, prominentes, o en forma de asa; labio leporino/paladar hendido o paladar ojival (fig. 1) y anomalías dentales. Si la talla es normal al nacer, los neonatos presentan pronto un retraso en el crecimiento de gravedad variable como el que presentaba la paciente descrita. La microcefalia por su parte no es constante. Las anomalías musculoesqueléticas incluyen: braquidactilia del 5.° dedo, braquimesofalangia, clinodactilia del 5.° dedo, anomalías de la columna vertebral e hipermovilidad y dislocación articulares. Otro signo cardinal del SK son las anomalías de los dermatoglifos con persistencia del almohadillado fetal de los dedos como presentaba el caso descrito.
Casi todos los pacientes tienen un déficit intelectual entre leve y moderado y pueden presentar manifestaciones neurológicas como hipotonía o convulsiones. Es frecuente un retraso madurativo global junto con una pérdida de audición que puede tener una causa neurosensorial o ser consecuencia de una otitis media crónica debida a malformación craneofacial o susceptibilidad a las infecciones como ocurría en la paciente. Los hallazgos oculares son ocasionales. Son frecuentes los defectos congénitos del corazón como las lesiones obstructivas del lado izquierdo o los defectos septales. Las anomalías renales y del conducto urinario son menos frecuentes, pero están presentes en aproximadamente un 25% de los pacientes con SK. En las niñas, la telarquia prematura puede ocurrir, pero no requiere tratamiento a menos que existan otros signos de pubertad precoz. También se ha documentado, sobre todo en adolescentes, disfunción inmune asociado a trombocitopenia, motivo por el cual presentamos este caso5.
El diagnóstico diferencial del SK incluye los síndromes: CHARGE, branquio-oto-renal, Ehlers-Danlos (forma hipermóvil) y Hardikar, los trastornos ligados a IRF6 y el síndrome de microdeleción 22q11. Diversas anomalías cromosómicas pueden producir signos que se solapan con el espectro clínico del SK9.
Aunque la morbilidad es significativa, el pronóstico es bastante favorable y la esperanza de vida depende en gran parte de las complicaciones cardiacas e inmunológicas. Por todo ello, resulta fundamental la aportación del laboratorio clínico en la orientación y seguimiento de esta entidad permitiendo así un estrecho control de sus complicaciones asociadas9.
