


La preeclampsia se define como la aparición de hipertensión y proteinuria a partir de la semana 20 de gestación. Afecta al 3-10% de las gestaciones en todo el mundo y se asocia a una importante morbimortalidad tanto materna como fetal. Aunque en la fisiopatología de la preeclampsia intervienen diversos factores, el más importante es la instauración de una insuficiencia placentaria. Esta es responsable de la inducción de un estado antiangiogénico en la gestante y del desarrollo de una disfunción endotelial en diversos órganos que desencadena las manifestaciones clínicas de la enfermedad. En los últimos años los criterios diagnósticos han sido actualizados y se ha propuesto el uso de nuevos marcadores, como el ácido úrico o los factores reguladores de la angiogénesis. Estas nuevas herramientas permiten un diagnóstico rápido y un manejo clínico adecuados, que son cruciales para minimizar el desarrollo de complicaciones.
Preeclampsia is defined by the onset of hypertension and proteinuria after 20 weeks gestation. It affects 3-10% of pregnancies worldwide and it is associated to a high morbidity and mortality, both for the mother and the fetus. Although several factors are involved in the physiopathology of preeclampsia, placental insufficiency is the most important of them. This is responsible for the induction of an anti-angiogenic state in the mother and the development of endothelial dysfunction in several organs, resulting in the clinical manifestations of the disease. In recent years the diagnostic criteria have been updated and the use of new biomarkers of the disease, mainly uric acid or angiogenesis related factors, have been proposed. These tools allow quick diagnosis and proper clinical management, which are crucial to minimize the development of complications.
La preeclampsia (PE) es un trastorno hipertensivo del embarazo, definido como la aparición de novo de hipertensión arterial y proteinuria a partir de la semana 20 de gestación1. En función de la edad gestacional a la que se instaura, se puede diferenciar entre PE precoz (antes de 34 semanas) y PE tardía (a las 34 semanas o posteriormente). La relevancia de esta clasificación va más allá de etiquetar la enfermedad, ya que estos subtipos difieren en su fisiopatología, complicaciones derivadas y manejo clínico.
EpidemiologíaLa PE complica el 3-10% de las gestaciones, aunque la falta de estandarización en cuanto a su diagnóstico y la heterogeneidad de las poblaciones estudiadas dificultan las estimaciones2. La incidencia de la enfermedad es superior en los países en vías de desarrollo, aunque en los últimos años se ha descrito un incremento en el número de casos de PE en países industrializados2–4. Por ello, la PE constituye un importante problema de salud a nivel global, más aún si consideramos su elevada morbimortalidad, tanto materna como fetal. De este modo, un reciente estudio reveló que en EE. UU., entre 2006 y 2010, el 8,9% de las muertes maternas tuvieron como causa la PE o la eclampsia5. Además se considera que una cuarta parte de las muertes perinatales en los países desarrollados son consecuencia de la PE/eclampsia, cifras aún más elevadas en los países con menos recursos6.
Se han descrito diversos factores que aumentan el riesgo de desarrollar la enfermedad. La PE se considera una enfermedad asociada al primer embarazo, y de hecho el riesgo es hasta 3 veces superior en gestantes nulíparas que en las multíparas7. Sin embargo, cuando el segundo o posteriores embarazos se producen con una pareja diferente, el efecto protector de la multiparidad desaparece8. Este hecho muestra la implicación de factores de origen inmunológico, una hipótesis que también explicaría por qué aumenta el riesgo de PE en embarazos logrados mediante técnicas de reproducción asistida, más aún cuando se utilizan gametos de donante9,10. El riesgo es especialmente elevado en gestantes con historia previa de PE, y aunque en menor medida, también cuando los antecedentes se presentan en familiares de primer grado, ya sean de la propia gestante o del padre, lo que indica la existencia de una base genética11. Además, se ha evidenciado una mayor incidencia y gravedad de la enfermedad en gestaciones múltiples, especialmente cuando son monocoriónicas12. Factores de riesgo de menor peso pero que también deben considerarse son la edad materna extrema7 o la etnia afroamericana13. Por otra parte, la presencia de otras condiciones clínicas subyacentes como sobrepeso14, hipertensión crónica15, insuficiencia renal16, diabetes mellitus17, enfermedades autoinmunes (como síndrome antifosfolípido)18 o algunas trombofilias19, predispone al desarrollo de la enfermedad.
FisiopatologíaLa PE no es una enfermedad aislada, sino que debe considerarse un síndrome multiorgánico con origen en la placenta. En la fisiopatología de la enfermedad pueden distinguirse 2 etapas: una primera etapa, que transcurre entre el primer y segundo trimestres, en la que se instaura una disfunción placentaria; y una segunda etapa, a partir del tercer trimestre, en la que se produce la respuesta materna a dicha disfunción.
Primera etapa: Disfunción placentariaEn el proceso fisiológico de la placentación, un grupo de células placentarias, los citotrofoblastos extravellosos, adquieren capacidad invasiva y migran hacia la decidua y primer tercio del miometrio materno, donde participan en el remodelado de las arterias espirales. En concreto, algunos citotrofoblastos se transforman en células con fenotipo endotelial, sustituyen al endotelio original materno y modifican el estroma, volviéndolo más laxo. Como consecuencia las arterias espirales se convierten en vasos sanguíneos con un diámetro mayor y que oponen una menor resistencia al flujo sanguíneo, lo que asegura una perfusión placentaria adecuada para el desarrollo normal del feto20,21. Sin embargo, en la PE los citotrofoblastos muestran una capacidad invasiva limitada y mantienen el fenotipo progenitor, por lo que no alcanzan el miometrio y no son capaces de transformar el endotelio materno. De este modo, las arterias espirales mantienen un calibre reducido y una resistencia aumentada, limitando el flujo sanguíneo desde la madre al feto21,22. La isquemia resultante provoca lesiones en la placenta (formación de nudos sincitiales, necrosis, fibrosis, eritroblastosis) que acentúan la disfunción placentaria23,24.
Con respecto a cuál es la causa primaria que origina la alteración de la placentación, se ha planteado la contribución de factores de diversa etiología. En primer lugar, se considera la existencia de una predisposición genética, de modo que se han identificado varios genes asociados con la enfermedad. Estos genes, de origen tanto materno como fetal, desarrollan funciones importantes durante la placentación (crecimiento y diferenciación celular, interacción intercelular, regulación de la respuesta inmunológica, entre otras), por lo que la presencia de determinados polimorfismos puede alterar este proceso25.
Por otro lado, se han descrito alteraciones del sistema inmunológico que podrían estar implicadas en el desarrollo de la enfermedad. De este modo se ha descrito que las células natural killer uterinas que participan como reguladoras de la placentación muestran un fenotipo aberrante en gestantes con PE, lo que puede impedir una interacción adecuada con los citotrofoblastos y bloquear el remodelado de las arterias espirales26,27. Además, durante la gestación el organismo favorece la proliferación de linfocitos con actividad reguladora generando un «estado inmunológico tipo Th2», lo que asegura una cierta inmunosupresión que evita la reacción materna frente a los antígenos fetales. Sin embargo, en la PE se ha demostrado un aumento de linfocitos Th1 y Th17, subpoblaciones que mediante la producción de citocinas inducen un estado proinflamatorio que favorece la incompatibilidad maternofetal28,29. En algunos casos, también se ha demostrado una relación entre la enfermedad y la producción de autoanticuerpos capaces de activar el receptor de angiotensina ii tipo 1, lo que provoca hipertensión y agrava la respuesta inflamatoria30.
Finalmente, factores ambientales también desempeñan su papel. Así, un elevado estrés oxidativo puede provocar lesiones placentarias y, en combinación con un estado proinflamatorio e hiperlipidemia, puede contribuir a la aterosis de las arterias espirales y acentuar el bloqueo del flujo sanguíneo31,32.
Etapa 2: Inducción de un estado antiangiogénicoLa reducción de la perfusión placentaria provoca fluctuaciones en la tensión de oxígeno, dando lugar a una sucesión de fases de hipoxia y de reperfusión que agravan el estrés oxidativo y la respuesta inflamatoria. Como consecuencia se desencadena una alteración en la producción de factores de origen placentario, de manera que disminuyen los factores con actividad angiogénica, como el factor de crecimiento del endotelio vascular A (VEGF-A) o el factor de crecimiento placentario (PlGF); mientras que aumentan los que presentan actividad antiangiogénica, como la tirosina cinasa tipo fms 1 soluble (sFlt-1) o la endoglina soluble33–35. En un embarazo normal, VEGF-A y PlGF estimulan la angiogénesis y regulan la función endotelial a través de la unión a sus receptores específicos, entre ellos el Flt-136. En la PE el exceso de sFlt-1, que es la forma truncada del receptor Flt-1, actúa como antagonista del VEGF-A y PlGF circulantes. De este modo los secuestra y bloquea su actividad, induciendo un estado antiangiogénico en la gestante. De acuerdo con esta teoría, se ha demostrado que el sFlt-1 bloquea la angiogénesis en modelos in vitro e induce hipertensión arterial, proteinuria y disfunción endotelial cuando es administrado a ratas preñadas37,38, daños que revierten cuando se aporta VEGF de manera exógena38. Asimismo, se ha descrito que pacientes oncológicos que reciben terapia con anticuerpos anti-VEGF (lo que ejercería un efecto análogo al del exceso de sFlt-1) desarrollan manifestaciones clínicas semejantes a las de gestantes con PE39. Por otro lado, la endoglina soluble podría actuar inhibiendo la vasodilatación al interferir con el óxido nítrico o con la endotelina-1, de forma independiente o bien magnificando el efecto del sFlt-140,41.
El balance antiangiogénico en conjunto con el resto de factores involucrados (predisposición materna, estrés oxidativo, respuesta inflamatoria, presencia de autoanticuerpos, hiperlipidemia, etc.) contribuye a una disfunción endotelial que en última instancia es responsable de las manifestaciones clínicas de la PE: hipertensión, proteinuria, disfunción hepática, alteraciones neurológicas y alteraciones hematológicas.
Aunque el modelo propuesto pretende simplificar la fisiopatología de la PE, hay que destacar que la etiología es multifactorial y que los desencadenantes de la enfermedad pueden ser diferentes en cada caso. De hecho existen importantes matices que difieren en la PE precoz y tardía, hasta tal punto que han llegado a considerarse entidades clínicas diferentes. En concreto, la PE precoz se caracteriza por un mayor número de lesiones placentarias y una alteración más grave en la producción de factores reguladores de la angiogénesis. Por otro lado, la PE tardía se asocia a una disfunción placentaria más leve, de modo que otros factores de naturaleza materna (obesidad, hipertensión crónica, etc.) son claves en el desarrollo de la enfermedad42–44.
Cuadro clínicoLa hipertensión arterial es una de las principales características de las gestantes con PE. Esta se produce como consecuencia de la alteración en la producción de factores reguladores del tono vascular, del incremento de la resistencia vascular y de la inducción de la vasoconstricción debida a la disfunción endotelial45.
Otro hallazgo frecuente es la alteración de la función renal, que es la responsable de la proteinuria. Las gestantes con PE presentan una lesión renal característica conocida como endoteliosis glomerular, que implica aumento del volumen glomerular, estrechamiento y oclusión de la luz de los capilares, presencia de depósitos de fibrina y pérdida de las fenestraciones de las células endoteliales46. Además, los podocitos muestran importantes alteraciones y una mayor tasa de apoptosis, lo que compromete aún en mayor medida la integridad de la barrera de filtración renal47.
Especialmente en los casos de mayor gravedad pueden observarse alteraciones a nivel hepático, lo que explica por qué en muchos casos uno de los síntomas referidos por las gestantes con PE es el dolor epigástrico. Estas alteraciones son la consecuencia de la disfunción endotelial de los sinusoides hepáticos, que desencadena fibrosis, trombosis y en algunos casos necrosis del parénquima hepático48. De hecho, la disfunción hepática es uno de los criterios del síndrome de HELLP (hemólisis, elevación de enzimas hepáticas, disminución de plaquetas), una complicación de la PE definida por el desarrollo de hemólisis intravascular (lactato deshidrogenasa≥600U/L), elevación de transaminasas hepáticas (aspartato-aminotransferasa≥70U/L) y trombocitopenia (recuento de plaquetas≤100.000/μL)]49.
Las alteraciones neurológicas son frecuentes, y se manifiestan como dolor de cabeza, fotopsias o, en el caso de la eclampsia, como convulsiones. Aunque se desconocen los mecanismos exactos, se ha planteado que la disfunción endotelial y la hipertensión arterial características de la PE provocan alteraciones en la función de la vasculatura cerebral, de modo que se produce una reducción del flujo sanguíneo, edemas o incluso infartos del tejido nervioso en los casos más graves50.
A nivel hematológico es habitual encontrar anemia hemolítica y/o trombocitopenia, 2 signos que son característicos del síndrome de HELLP51. En algunos casos se produce una activación descontrolada del sistema de la hemostasia, lo que puede culminar con el desarrollo de coagulación intravascular diseminada52.
La alteración de la permeabilidad vascular provoca la aparición de edemas en las gestantes que en los casos más graves pueden llegar a desarrollar edema pulmonar51.
La aparición de las complicaciones mencionadas no es común a todos los casos de PE y depende de varios factores. Entre ellos, destacan la edad de presentación clínica (la probabilidad de complicaciones es mayor en la PE precoz), la gravedad del proceso, el manejo clínico adecuado o la presencia de otras enfermedades subyacentes, como la hipertensión crónica53,54.
Criterios diagnósticosLos criterios clásicamente utilizados en el diagnóstico de la PE son los mismos que definen la enfermedad: hipertensión arterial y proteinuria55.
- a)
Hipertensión arterial: definida como tensión arterial sistólica≥140mmHg y/o tensión arterial diastólica≥90mmHg, medida en 2 ocasiones separadas al menos por 4h, a partir de las 20 semanas de gestación en una mujer sin hipertensión previamente diagnosticada; o bien una tensión arterial sistólica≥160mmHg y/o tensión arterial diastólica≥110mmHg, medida en 2 ocasiones separadas por unos minutos.
- b)
Proteinuria: definida como la excreción de≥300mg de proteína en orina de 24h; o un cociente proteína/creatinina≥0,3 (medidas ambas en mg/dL). Debido a la variabilidad de los métodos cualitativos, se desaconseja la determinación de proteinuria con tiras reactivas, salvo en ausencia de métodos cuantitativos disponibles, en cuyo caso se consideraría proteinuria a partir de un valor de 1+ en proteínas.
Sin embargo, dado que estos criterios no cubren el espectro global de PE, recientemente han sido actualizados56, de modo que en ausencia de proteinuria, se admite que existe PE cuando aparecen hipertensión arterial y algún indicativo de disfunción orgánica materna, entendida como uno de los siguientes:
- -
Trombocitopenia (recuento de plaquetas≤100.000/μL).
- -
Insuficiencia renal (concentración sérica de creatinina≥1,1mg/dL o incremento de la creatinina sérica del doble, en ausencia de otra enfermedad renal).
- -
Alteración de la función hepática (concentración sérica de transaminasas hepáticas del doble sobre el valor normal).
- -
Edema pulmonar.
- -
Síntomas cerebrales o visuales.
Los criterios diagnósticos así como los signos y síntomas clínicos que presentan las gestantes con PE muestran importantes limitaciones. Muchos de estos parámetros son inespecíficos e incluso subjetivos, no siempre correlacionan con la gravedad de la enfermedad, no se anticipan a la instauración de la misma y no permiten realizar el diagnóstico en gestantes con hipertensión o proteinuria previas, así como en los casos de PE atípica, es decir, aquellos en los que los criterios diagnósticos clásicos no están presentes. Por ello, se han destinado múltiples esfuerzos a la identificación de nuevos marcadores de laboratorio que superen estos obstáculos, algunos de los cuales se detallan a continuación.
PlaquetasEl recuento de plaquetas es clave en la evaluación de la gestante con PE, ya que la trombocitopenia es uno de los signos de gravedad de la PE y criterio diagnóstico del síndrome de HELLP49,56. Aunque recuentos de plaquetas bajos se han relacionado con un mayor riesgo de desarrollar PE57 el uso de este marcador aislado no ha demostrado un rendimiento aceptable en el diagnóstico de la enfermedad, con áreas bajo las curvas Receiver Operating Characteristic entre 0,42-0,69 según los estudios57–59. Por otra parte, aunque un recuento de plaquetas disminuido se ha relacionado con un mayor riesgo de complicaciones maternas, su baja sensibilidad (16-33%) limita su aplicación como marcador pronóstico60.
TransaminasasDado que las gestantes con PE muestran en ocasiones alteraciones de la función hepática, se ha planteado la utilización de las enzimas aspartato-aminotransferasa y alanina-aminotransferasa en el diagnóstico de la enfermedad. De hecho, la elevación de estas enzimas es uno de los indicadores de PE con criterios de gravedad56 y criterio diagnóstico del síndrome de HELLP49. Sin embargo, la principal limitación de estas pruebas es su baja sensibilidad, y es que se ha descrito que solo se alteran en el 10% de las gestantes con PE48. Del mismo modo, la elevación de las pruebas de función hepática se relaciona con el desarrollo de complicaciones maternas y fetales derivadas de la PE, si bien, debido a la baja sensibilidad, la presencia de valores dentro del rango de referencia no permite excluir el riesgo de dichas complicaciones61.
Ácido úricoLas gestantes con PE presentan concentraciones séricas de ácido úrico superiores a las de mujeres con embarazos sin complicaciones. Por este motivo este marcador bioquímico ha sido propuesto en el diagnóstico de la enfermedad y de hecho se utiliza habitualmente en la valoración de la gestante con sospecha de PE62. Aunque existe cierta heterogeneidad en los puntos de corte evaluados en los diferentes estudios, en general el ácido úrico ha mostrado mayor rendimiento diagnóstico que las pruebas de función hepática63. De este modo, alteraciones en los niveles de ácido úrico permiten diagnosticar PE, especialmente cuando es precoz, con una especificidad muy elevada (en torno al 95% en los estudios más optimistas) pero tal y como ocurría con las transaminasas, valores de este parámetro dentro del rango de referencia no permiten excluir la enfermedad debido a su moderada sensibilidad, que se sitúa entre el 60-87% según las series analizadas64,65. Por otra parte, el ácido úrico ha demostrado ser útil en el pronóstico de complicaciones derivadas de la PE, tanto maternas como fetales66. Además, se ha evidenciado la existencia de una correlación negativa entre la concentración de ácido úrico en el momento de la admisión en urgencias y los días de manejo clínico expectante, por lo que este marcador podría ser útil en la predicción de parto inminente contribuyendo así a la optimización en el manejo de estas gestantes67.
El hecho de que algunos estudios hayan demostrado que la hiperuricemia en gestantes con PE precede a la instauración de la enfermedad (incluso ya en el primer trimestre) puede indicar que el ácido úrico desempeña un papel en la fisiopatología de la enfermedad68,69. En consonancia con este planteamiento, se ha descrito que el ácido úrico inhibe la invasión trofoblástica y el remodelado de las arterias uterinas in vitro, procesos que precisamente se desarrollan en la primera etapa de la gestación70. La hiperuricemia también ha sido relacionada con la activación de la respuesta inflamatoria y con el estrés oxidativo, factores asociados a la enfermedad71–73. Sin embargo, esta hipótesis contrasta con los resultados publicados recientemente por Chen et al.74, que en un amplio estudio prospectivo no fueron capaces de demostrar una asociación entre la PE y los niveles de ácido úrico medidos en el primer o segundo trimestre de gestación. En cambio, la concentración de ácido úrico en el momento de la presentación clínica sí que estuvo asociada con el desarrollo de la enfermedad, por lo que consideraron que la hiperuricemia en estas gestantes era secundaria a la alteración renal inducida por la PE, que impedía una excreción adecuada del metabolito.
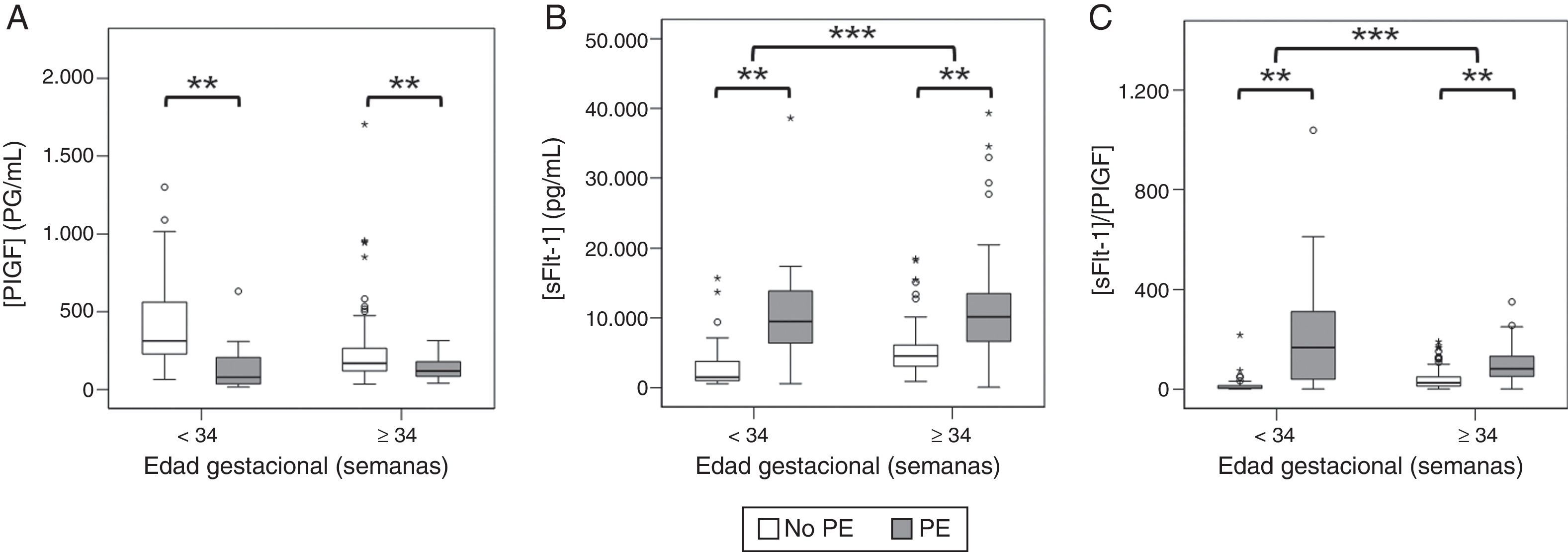
Marcadores reguladores de la angiogénesisEn las últimas décadas, numerosos estudios han evaluado la utilidad de los marcadores reguladores de la angiogénesis en el diagnóstico de PE75–79. La mayor parte del interés se ha centrado en los factores PlGF y sFlt-1, y en especial, en el cociente entre ambos (sFlt-1/PlGF), cuyo rendimiento ha resultado ser mayor que el de cualquiera de las otras 2 determinaciones aisladas (fig. 1)76. La capacidad diagnóstica de estos marcadores ha demostrado ser superior en la PE precoz que en la tardía65,76. Este hecho puede explicarse porque la alteración en la producción de PlGF y sFlt-1 es el reflejo directo de la disfunción placentaria, que como se mencionó previamente, es mayor en la PE precoz. Precisamente es en este subgrupo de gestantes donde resulta más importante identificar la enfermedad, ya que son las que revisten mayor gravedad y tienen un riesgo más elevado de desarrollar complicaciones53,80.
, sFlt-1 (B) y cociente sFlt-1/PlGF (C) en gestantes con signos y/o síntomas de PE. Las gestantes que posteriormente desarrollaron PE precoz (izquierda) o tardía (derecha) mostraron valores más bajos de PlGF y más altos de sFlt-1 y del cociente sFlt-1/PlGF que las que no desarrollaron la enfermedad. ** p<0,001; *** p<0,05. Modificado de Álvarez-Fernández et al.61.")
Concentración de PlGF (A), sFlt-1 (B) y cociente sFlt-1/PlGF (C) en gestantes con signos y/o síntomas de PE. Las gestantes que posteriormente desarrollaron PE precoz (izquierda) o tardía (derecha) mostraron valores más bajos de PlGF y más altos de sFlt-1 y del cociente sFlt-1/PlGF que las que no desarrollaron la enfermedad. ** p<0,001; *** p<0,05. Modificado de Álvarez-Fernández et al.61.
Sin embargo, aún existe una cierta controversia con respecto a los puntos de corte que deben utilizarse para estos marcadores76,77,81,82, por lo que recientemente se ha publicado un documento de consenso que pretende estandarizar su uso83:
- -
Un cociente sFlt-1/PlGF>85 (PE precoz) o>110 (PE tardía) indica una elevada probabilidad de desarrollar PE u otra forma de insuficiencia placentaria (retraso del crecimiento intrauterino, desprendimiento prematuro de placenta). Estos puntos de corte han demostrado una elevada especificidad (>95%) para el ensayo Elecsys (Roche)81, un resultado que ha sido reproducido recientemente con el ensayo también electroquimioluminiscente de KRYPTOR (BRAHMS)84.
- -
Un cociente sFlt-1/PlGF<38 excluye el desarrollo de PE, al menos en un plazo inferior a una semana, con un valor predictivo negativo del 99%, de acuerdo con un reciente estudio prospectivo que también empleó el ensayo Elecsys (Roche)82.
- -
En base a estos 2 estudios llevados a cabo con el ensayo de Roche81,82, un cociente entre 33-85 (PE precoz) o 33-110 (PE tardía) indica una baja probabilidad de padecer la enfermedad en el momento de análisis, pero un riesgo de desarrollarla en un plazo de 4 semanas.
Además, el cociente sFlt-1/PlGF puede ser indicador de la gravedad de la PE, de manera que valores extremadamente elevados (>655 o>201 para la PE precoz o tardía, respectivamente) se han asociado con la necesidad de inducir el parto en menos de 48h83,85,86. Otros estudios también han hallado una correlación negativa entre los valores del cociente sFlt-1/PlGF y el tiempo transcurrido desde la determinación hasta que se produjo el parto, independientemente de que las gestantes desarrollaran PE o no65,79. De este modo, este marcador puede ser utilizado para identificar a las gestantes que tienen un mayor riesgo de complicaciones y que requieren una monitorización más estrecha así como medidas terapéuticas específicas. En todo caso se necesitan estudios prospectivos que confirmen la utilidad de este marcador en la práctica clínica y que validen los puntos de corte propuestos con diferentes ensayos analíticos.
Manejo clínicoPrevenciónEl primer paso en la prevención de la PE implica identificar a la población que presenta riesgo de desarrollar la enfermedad. En los últimos años se han planteado numerosos modelos de predicción precoz de PE que combinan características maternas, marcadores bioquímicos e índices ecográficos, aunque con resultados muy diferentes87–90. Recientemente, Poon y Nicolaides91 han propuesto un modelo de cribado de PE precoz en primer trimestre que permitiría alcanzar una tasa de detección del 96% para una tasa de falsos positivos del 10%, utilizando 4 parámetros: tensión arterial media, índice de pulsatilidad de arterias uterinas, concentración de PlGF y de la proteína plasmática A asociada al embarazo. Sin embargo, estos resultados aún no han sido reproducidos por otros grupos, por lo que se requieren más estudios antes de que un programa de este tipo pueda ser implantado en la práctica clínica. Por este motivo, actualmente la prevención se dirige a población de riesgo en base a características maternas (factores de riesgo como historia previa de PE, gestaciones múltiples o condiciones clínicas subyacentes). En este tipo de población, se ha demostrado que la administración de aspirina en dosis bajas (60-80mg/día) antes de la semana 16 (cuando aún se está produciendo la transformación de las arterias espirales), reduce la incidencia de la enfermedad en un 29%, de parto pretérmino en un 19% y retraso del crecimiento intrauterino en un 20%92, por lo que esta medida profiláctica ya constituye una recomendación en la práctica clínica56,93.
TratamientoEl único tratamiento definitivo de la PE es la eliminación de la placenta, pero asegurando previamente el bienestar tanto materno como fetal. Es imprescindible la participación de un equipo multidisciplinar, que realice un seguimiento estrecho de la gestante mediante controles analíticos (hemograma, iones, función renal y hepática, coagulación, proteinuria) y valoración del bienestar fetal (ecografía, Doppler). Para ello, los casos más graves o precoces habitualmente requieren hospitalización, mientras que los casos más leves pueden seguir un control ambulatorio94.
Las intervenciones terapéuticas tienen como objetivo estabilizar la situación materna y evitar el desarrollo de complicaciones. De este modo, debe iniciarse tratamiento antihipertensivo (labetalol, hidralacina, alfametildopa) ante valores de tensión arterial sistólica≥160mmHg o diastólica≥110mmHg. Además, en casos de PE grave se recomienda la administración de sulfato de magnesio para prevenir o tratar las convulsiones de la eclampsia, tratamiento que debe mantenerse 24-48h posparto56,94.
La decisión de finalizar el parto implica sospesar entre el beneficio que supone para la gestante eliminar la placenta frente al riesgo de inducir un parto pretérmino. Se recomienda mantener una actitud expectante hasta las 37 semanas, y a partir de ese momento finalizar la gestación. Sin embargo, en casos graves puede ser necesaria una inducción más temprana. La recomendación general es esperar al menos hasta la semana 34, aunque un empeoramiento grave de la situación materna o fetal (hipertensión persistente, pródromos de eclampsia, edema pulmonar, retraso del crecimiento intrauterino grave, pérdida fetal) justifica la finalización antes de la semana 34, en cuyo caso se requiere la administración previa de corticosteroides para asegurar la maduración pulmonar fetal56,95.
Responsabilidades éticasDado que el presente manuscrito se trata de un artículo de revisión, no existen consideraciones éticas que declarar.
Conflicto de interesesDeclaramos que no existe conflicto de intereses.
