


Bajas concentraciones séricas de Cu en neonatos pueden ser la primera señal de una ingesta deficiente de este elemento o, alternativamente, de enfermedades genéticas que afectan su metabolismo. Desgraciadamente, es difícil la interpretación de las concentraciones de Cu en esta población, ya que están influenciadas por distintos factores, entre ellos la prematuridad, el tipo de alimentación y la presencia de un estado inflamatorio. Sin embargo, en el caso que aquí se describe fue la baja concentración sérica de Cu la primera pista para el diagnóstico de enfermedad de Menkes. Se demuestra así la utilidad de la determinación de Cu dentro de protocolos neurometabólicos y de retraso psicomotor en población neonatal y lactante.
Low serum Cu concentrations in newborns can be the first indication of a severe Cu deficient intake or, alternatively, of genetic diseases affecting Cu metabolism. Unfortunately, interpretation of serum Cu concentrations in this population is difficult because they also influenced by several variables, such as, prematurity, type of feeding and inflammatory conditions. However, in the case described in this paper was a low serum Cu concentration the first clue for diagnosing Menkes disease. It is so demonstrated the usefulness of Cu determination within neurometabolic or psychomotor retardation protocols for newborn and infant populations.
Niño nacido a las 38 semanas de gestación tras un embarazo bien controlado y parto eutócico. Al nacer pesó 3.190g y tuvo un Apgar de 10/10. El cribado neonatal metabólico-endocrino no mostró alteraciones (29 enfermedades cribadas).
Con 28 días de vida acude a su pediatra por dificultad con las tomas, habiendo ganado solo 100g desde el nacimiento. Los padres refieren que han notado la piel arrugada desde los primeros días. En la anamnesis se objetiva cierto aspecto distrófico, retrognatia, orejas de implantación baja y descamación cutánea, por lo que se ingresa para efectuarle un estudio. La analítica de sangre no presentó alteraciones significativas y las ecografías transfontanelar y abdominal (incluyendo renal) fueron normales. El cariotipo también resultó normal (46,XY). Durante el ingreso presentó febrícula de manera intermitente, que cedía espontáneamente, y episodios de desaturación con cianosis generalizada y congestión nasal, por lo que se realizó aspirado nasofaríngeo resultando positivo a gripe B, requiriendo el aporte suplementario de oxígeno con gafas nasales para mantener saturaciones adecuadas. Paulatinamente presenta mejoría clínica, realiza mejor las tomas y desaparecen los episodios de cianosis, por lo que se da de alta (40 días).
A los 56 días de vida en el control por su pediatra se constata de nuevo rechazo en las tomas, estancamiento ponderal, náuseas y vómitos de una semana de evolución, siendo remitido a urgencias del hospital de referencia. Los padres refieren que no consiguen sonrisa social y que los últimos días notan pies y manos frías, lo que no habían advertido previamente. Por el contrario, sí existe seguimiento visual, reconociendo a los padres.
Se ingresa y se realiza estudio de retraso psicomotor. Se diagnostica e interviene de estenosis hipertrófica de píloro. En las analíticas realizadas según protocolo destaca un Cu y ceruloplasmina (Cp) muy bajos: Cu=6,0μg/dl (18-94), Cp=2,2mg/dl (6,1-34,5), pero dados los antecedentes de mala ingesta y vómitos, se decide control analítico evolutivo para decidir la actitud a seguir.
Tres semanas después (77 días) se valora en consultas externas de neurometabolismo. A la exploración física destaca escafocefalia marcada, pabellones auriculares puntiagudos, mejillas prominentes, piel muy blanca y pelo deslustrado y despigmentado, aunque no se puede valorar si existe pili torti. Respecto a la exploración neurológica presenta un llanto vigoroso e hipotonía axial con rezagamiento cervical y discreto seguimiento visual. Sigue sin mostrar sonrisa social. Las concentraciones séricas de Cu y Cp se mantienen bajas, por lo que se sospecha enfermedad de Menkes (EM) y se solicita estudio genético, radiológico y encefalograma (EEG).
A los 3 meses de vida el EEG no muestra grafoelementos patológicos. Sin embargo, en la radiografía de cráneo se observan huesos wormianos en suturas sagital y lambdoideas, y prominencia de partes blandas en la región frontal. En la radiografía de fémur bilateral se aprecia ensanchamiento metafisario y espolones óseos en metáfisis proximal y distal de ambos fémures. Puesto que los hallazgos radiológicos concuerdan con la sospecha clínica (pendiente de confirmación genética) se inicia tratamiento con histidinato de cobre a dosis de 100μg /kg administrados diariamente por vía subcutánea.
Un mes después (4 meses) la madre refiere que realiza movimientos en sacudidas. Se realiza un EEG que muestra un trazado con deficiente estructuración y mala diferenciación de la actividad eléctrica cortical. Al mes siguiente (5 meses) se confirma en el EEG el registro anormal por la asimetría de la actividad de fondo, así como por la presencia de grafoelementos de morfología aguda que son más frecuentes que en el estudio previo, indicando empeoramiento, y se inicia tratamiento antiepiléptico con ácido valproico.
A los 6 meses de vida se recibe el informe genético que confirma la presencia de la mutación sin sentido p.R986X (p.Arg256Ter) en la posición 2956 (c.2956C>T) del exón 15 del gen ATP7A, lo que provoca que la proteína ATPasa 7A sea más corta, con solo 985 aminoácidos frente a los 1.500 de la proteína normal. Esta mutación ha sido descrita previamente como asociada al desarrollo de EM1.
Actualmente (7 meses) el niño permanece en seguimiento, siendo difícil valorar la efectividad del tratamiento, aunque parece presentar una evolución clásica de la enfermedad.
DiscusiónEl Cu es un elemento esencial que participa en multitud de procesos vitales para el organismo2. En el plasma aproximadamente el 75-90% del Cu está unido a la Cp, de forma que los niveles séricos de Cu evolucionan de forma paralela a los de Cp. Así, las situaciones de hipoproteinemia severa (malnutrición, malabsorción, cirrosis, síndrome nefrótico) disminuirán las concentraciones séricas de Cp (y por tanto de Cu), mientras que las situaciones de inflamación, infección y los estrógenos (embarazo o anovulatorios) incrementan notablemente sus concentraciones3.
La detección temprana de concentraciones bajas de Cu (y/o Cp) en neonatos/lactantes puede ser útil para identificar una ingesta insuficiente de Cu (lo que puede ocurrir con fórmulas artificiales muy deficitarias en Cu)4 o enfermedades genéticas relacionadas con el Cu (como la enfermedad de Wilson [EW] o la EM)5. Desafortunadamente, esta población muestra un amplio rango de concentraciones de Cu, lo que conduce a una superposición de valores entre niños sanos y afectos. Muchos bebés muestran de forma fisiológica unos bajos niveles de Cu sérico, mientras que en otros pueden estar incrementados asociados a ciertas enfermedades6.
Recientemente, en un estudio con neonatos hospitalizados, hemos demostrado que el factor más importante que modifica sus concentraciones séricas de Cu es la presencia de un estado inflamatorio7. La Cp es una proteína de fase aguda que incrementa su concentración durante la inflamación y, como consecuencia, se produce una elevación concomitante de Cu8.
Otro factor a considerar es el grado de prematuridad. Los neonatos de muy bajo peso suelen tener valores séricos de Cu muy disminuidos8. El Cu se acumula en el hígado fetal en el tercer trimestre de gestación, actuando como fuente de reserva, y tras el parto su concentración en suero se va incrementando de forma progresiva hasta alcanzar niveles similares a los de adultos aproximadamente a los 6 meses de vida9.
Otros factores que también modifican las concentraciones de Cu en neonatos, aunque en menor medida que los anteriores, son el tipo de alimentación (materna o artificial), los días de vida en la extracción y la posible suplementación de cinc en la madre o en el neonato7.
Por todo ello surgen dudas sobre la utilidad clínica de la determinación de Cu (y/o Cp) en el periodo neonatal/lactante, ya que en muchas ocasiones es difícil interpretar los resultados obtenidos. Así, por ejemplo, diversos programas piloto para el cribado neonatal de la EW no han tenido éxito y se ha considerado que la edad ideal para su realización serían los 3 años de edad10. De la misma forma, deficiencias extremas de Cu en niños prematuros alimentados con fórmulas artificiales se detectaron cuando habían transcurrido varios meses4.
Además de los factores anteriores, también se ha de considerar la propia variabilidad introducida por la diferente metodología analítica disponible. Los programas externos de garantía de la calidad demuestran que la medición del Cu por métodos colorimétricos proporciona valores sensiblemente diferentes a los métodos atómicos, e incluso dentro de estos hay diferencias significativas entre, por ejemplo, la cámara de grafito y la llama. Por otra parte, la Cp sérica puede ser medida por métodos inmunoquímicos o por métodos enzimáticos. Estos últimos miden solo la actividad oxidasa, mientras que los primeros detectan la molécula de Cp y, por tanto, pueden cuantificar formas inactivas, sobrevalorando la concentración de Cp respecto de los métodos enzimáticos. Además, incluso dentro de los métodos inmunoquímicos se han observado grandes diferencias debido a una falta de estandarización de los ensayos3.
Además del Cu y la Cp, la estimación indirecta del Cu no unido a Cp ha sido otra magnitud que ha recibido cierto interés. Es un cálculo que hace uso de las concentraciones séricas de Cu y Cp y que considera una proporción constante de 6 átomos de Cu por molécula de Cp. A pesar de que esta estimación ha mostrado cierta utilidad en algunas situaciones, fundamentalmente como ayuda al diagnóstico de la EW cuando los valores de Cp y Cu están cerca de la normalidad, su uso de forma sistemática presenta numerosas limitaciones. En un porcentaje significativo de las muestras proporciona valores negativos, lo cual es teóricamente imposible. Esto es debido a que la relación Cu/Cp no es constante (por ejemplo, disminuye en el embarazo11) y a que el valor calculado depende ampliamente de la metodología analítica utilizada, a lo que hay que sumar la incertidumbre asociada a 2 determinaciones independientes (del Cu y Cp), cada una con su propia variabilidad intrínseca3.
Para tratar de superar todas las limitaciones enumeradas en los párrafos anteriores, durante los últimos años se ha propuesto el uso de nuevos biomarcadores de Cu7. Entre ellos están la medida del Cu sérico ultrafiltrable y/o intercambiable, la medida enzimática del Cu sérico no unido a Cp, el establecimiento de la relación isotópica 65Cu/63Cu en suero y/u orina o la cuantificación de péptidos específicos de la Cp a partir de muestras de sangre seca en papel de filtro digeridas con tripsina. Sin embargo, estos nuevos potenciales marcadores todavía no están completamente validados ni introducidos en la práctica clínica habitual. Por lo tanto, la determinación del Cu total sigue siendo la forma más accesible de evaluar el estado del metabolismo del Cu en un laboratorio clínico.
El caso que se presenta aquí demuestra que, al menos para el diagnóstico de la EM, sí que puede ser útil la inclusión de este parámetro en protocolos neurometabólicos y de retraso psicomotor en población neonatal/lactante, y que valores de Cu sérico por debajo de 20-25μg/dl deben levantar sospecha en el adecuado contexto clínico.
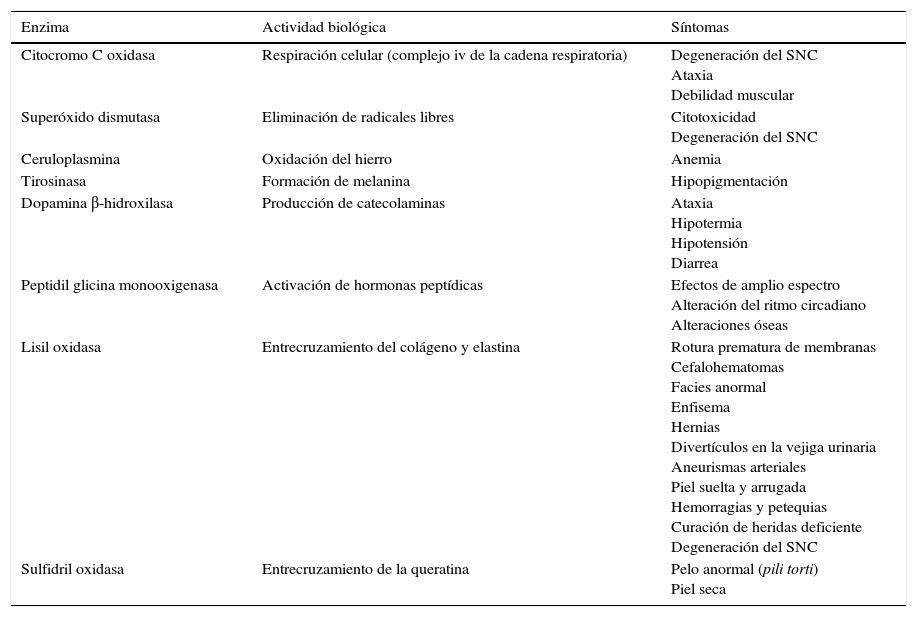
La EM es una enfermedad multisistémica letal del metabolismo del Cu que se transmite ligada al cromosoma X12. En consecuencia, la mayoría de los pacientes son varones, aunque se ha visto que mujeres portadoras pueden manifestar un grado variable de afectación13. Los varones con EM normalmente exhiben un curso muy grave, con muerte en los primeros meses/años de vida. Existe una forma más leve de la enfermedad que se denomina síndrome del asta occipital. En ambos casos el defecto se encuentra en el gen ATP7A que codifica la ATPasa 7A. Esta proteína transportadora de Cu se expresa en la mayoría de los tejidos (excepto en el hígado) y está involucrada en el transporte intracelular del Cu hacia la vía vesicular secretora, o en la exportación del exceso de Cu hacia fuera de la célula. Desempeña un papel decisivo en la absorción del Cu de la dieta por las células intestinales, y en la captación del Cu por el SNC facilitando su transporte direccional desde la sangre a las células neuronales a través de la barrera hematoencefálica. Los rasgos clínicos de la EM se relacionan con la actividad deficiente de las enzimas dependientes de Cu (tabla 1)12.
Principales enzimas dependientes de Cu y su relación con los síntomas de la enfermedad de Menkes
| Enzima | Actividad biológica | Síntomas |
|---|---|---|
| Citocromo C oxidasa | Respiración celular (complejo iv de la cadena respiratoria) | Degeneración del SNC Ataxia Debilidad muscular |
| Superóxido dismutasa | Eliminación de radicales libres | Citotoxicidad Degeneración del SNC |
| Ceruloplasmina | Oxidación del hierro | Anemia |
| Tirosinasa | Formación de melanina | Hipopigmentación |
| Dopamina β-hidroxilasa | Producción de catecolaminas | Ataxia Hipotermia Hipotensión Diarrea |
| Peptidil glicina monooxigenasa | Activación de hormonas peptídicas | Efectos de amplio espectro Alteración del ritmo circadiano Alteraciones óseas |
| Lisil oxidasa | Entrecruzamiento del colágeno y elastina | Rotura prematura de membranas Cefalohematomas Facies anormal Enfisema Hernias Divertículos en la vejiga urinaria Aneurismas arteriales Piel suelta y arrugada Hemorragias y petequias Curación de heridas deficiente Degeneración del SNC |
| Sulfidril oxidasa | Entrecruzamiento de la queratina | Pelo anormal (pili torti) Piel seca |
La EM comienza habitualmente en varones a los 2-3 meses de edad, con una regresión del desarrollo psicomotor asociada a la aparición de hipotonía, convulsiones y dificultad de medro. En la mayoría de ocasiones el pelo característico que presentan estos pacientes, que es corto, escaso, tosco y retorcido, junto con los síntomas anteriores, hacen sospechar la enfermedad14. Su observación al microscopio óptico revela la presencia de pili torti (giro de 180° del eje longitudinal). Otras manifestaciones habituales relacionadas con el tejido conectivo incluyen tortuosidad de los vasos sanguíneos, hernias umbilicales e inguinales, divertículos en la vejiga, pólipos gástricos, laxitud de piel y articulaciones y anomalías óseas.
El diagnóstico de la EM en la época neonatal es difícil, ya que las manifestaciones clínicas son sutiles y poco específicas. En ocasiones, la inusual pigmentación del pelo puede sugerir el diagnóstico, aunque en la mayoría de los casos el aspecto del pelo de los neonatos no llama la atención y la presencia de pili torti no es evidente. Neurológicamente, los neonatos con EM habitualmente parecen normales. En relación con la hipotermia, la EM es citada en Online Mendelian Inheritance in Man como la primera causa posible de hipotermia neonatal15.
La administración subcutánea o parenteral de histidinato de Cu parece ser efectiva en mejorar la esperanza y calidad de vida de los pacientes con EM, sobre todo cuanto más precozmente se instaura el tratamiento12. Sin embargo, no deja de ser una medida paliativa, ya que el Cu que alcanza las células de los tejidos no puede ser incorporado a las proteínas/enzimas de la vía secretora, y además el Cu administrado no puede alcanzar el SNC.
ConclusiónEl caso presentado demuestra la utilidad de la determinación del Cu en población neonatal/lactante para el diagnóstico de la EM ante síntomas poco específicos que pueden ser causados por múltiples enfermedades.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
