


Las hemoglobinopatías constituyen los trastornos monogénicos más frecuentes, sobre todo en determinadas razas y áreas, por su efecto protector frente a la malaria. Los cambios migratorios están provocando un aumento de estas alteraciones en el mundo occidental. La cromatografía líquida de alta resolución (HPLC) es el método de elección actual para la detección de hemoglobinopatías estructurales y cuantificación de hemoglobina A2 y fetal. Describimos un caso clínico donde se identificó una doble heterocigosis HbO-Arab y α-talasemia tras detección de microcitosis y una variante anómala de hemoglobina de menor valor del esperado, destacando la idoneidad del estudio multidisciplinar de este tipo de enfermedades.
Haemoglobinopathies are the most frequent monogenic disorders, particularly in certain races and areas, because of their protective effect against malaria. Migratory changes are leading to an increase in these conditions in the western world. High Performance Liquid Chromotography (HPLC) is nowadays a method of choice in detecting structural haemoglobinopathies and in the quantification of foetal and haemoglobin (Hb) A2. A clinical case is described in which a double heterozygous HbO-Arab and α-thalassaemia was identified following the detection of microcytosis and an anomalous haemoglobin variant, which was lower than expected – highlighting the appropriateness of a multidisciplinary study for these types of pathologies.
Las hemoglobinopatías constituyen un grupo heterogéneo de enfermedades hereditarias producidas por alteraciones de la molécula de hemoglobina (Hb); constituyen los trastornos monogénicos más frecuentes, sobre todo en determinadas razas y áreas, debido en parte a su efecto protector frente a la malaria1. Comúnmente se dividen en 2 tipos: hemoglobinopatías estructurales, por síntesis de una Hb anómala, y talasemias, por disminución o ausencia en la síntesis de una cadena normal. Los cambios migratorios en los últimos años han provocado un aumento de estas alteraciones en el mundo occidental2.
La Hb humana adulta es una mezcla de los subtipos HbA1 (α2β2) (normal >90%), HbA2 (α2δ2) (normal <3,5%) y Hb fetal (F) (α2γ2) (normal <1%). Hoy día la cromatografía líquida de alta resolución (HPLC), utilizada para la medición de la Hb glucosilada (HbA1c) en el diagnóstico y control de la diabetes mellitus, es el método de elección para la detección de hemoglobinopatías estructurales y la cuantificación de HbA2 y F3.
El gen de la α globina se localiza en el cromosoma 16 y cada individuo posee 2 cromosomas 16 (genotipo normal, αα/αα). Las α-talasemias se deben a la falta de síntesis de cadenas α, principalmente por deleción o pérdida de material genético. La deleción más frecuente en España, área mediterránea y población negra, es la que afecta a 3.7kb de ADN; la deleción de 4.2kb es más frecuente en el Sudeste asiático.
La HbO-Arab (también conocida como HbEgypt o HbO-Thrace) es una variante poco frecuente con funcionalidad y estabilidad normales. Se debe a una mutación puntual de la cadena β, en el codón 121, donde la sustitución del aminóacido ácido glutámico por lisina conlleva la sustitución de la base glicina por adenina (Glu>Lys; G>A). En el cromatograma de HPLC ocupa un lugar superponible al de la HbC. En estado heterocigoto no tiene repercusión clínica, con un 38-43% de Hb anómala; el estado homocigoto puede ser asintomático, con hemólisis compensada, o puede haber ictericia recurrente, anemia microcítica hipercrómica y esplenomegalia. Es relevante de cara al consejo genético por su posible combinación con HbS, que da lugar a una forma severa de enfermedad falciforme. Ha sido descrita en afroamericanos, en países balcánicos, en Egipto, en gitanos y en arábes4.
Material y métodosSe presenta el caso clínico de una mujer de 37 años, de raza árabe, natural de Marruecos, a la que se solicitó una primera analítica general desde atención primaria por sospecha de embarazo. El análisis del hemograma fue realizado en el sistema ADVIA 2120 (Siemens Diagnostics®). La detección de la variante de hemoglobinopatía estructural fue llevado a cabo con los sistemas ADAMSTM A1c HA-8160 y ADAMSTM A1c HA-8180V (A. Menarini Diagnostics®). Se trata de sistemas HPLC automatizados que usan cromatografía de intercambio catiónico de fase reversa para la separación de la HbA1c, F y HbA2 en programas de 4,2min el primero y 1,5 el segundo (252 y 90seg respectivamente). La absorbancia del eluido de Hb es cuantificada en una doble longitud de onda (415 y 500nm el primero, 420 y 500nm el segundo). Los estudios moleculares fueron llevados a cabo en laboratorios de referencia, usando técnicas en la primera muestra PCR multiplex (α-Globin StripAssayTM Vienna Lab® —21 deleciones y mutaciones— y β-Globin StripAssay MEDTM Vienna Lab® —22 mutaciones—), y en una segunda muestra de amplificación por PCR mediante primers específicos de los exones 1, 2 y 3 y de las regiones intrónicas adyacentes del gen de la cadena β, con secuenciación del fragmento amplificado y visualización de las secuencias mediante electroforesis capilar.
Los diversos análisis se han realizado tras obtener el consentimiento informado de la paciente.
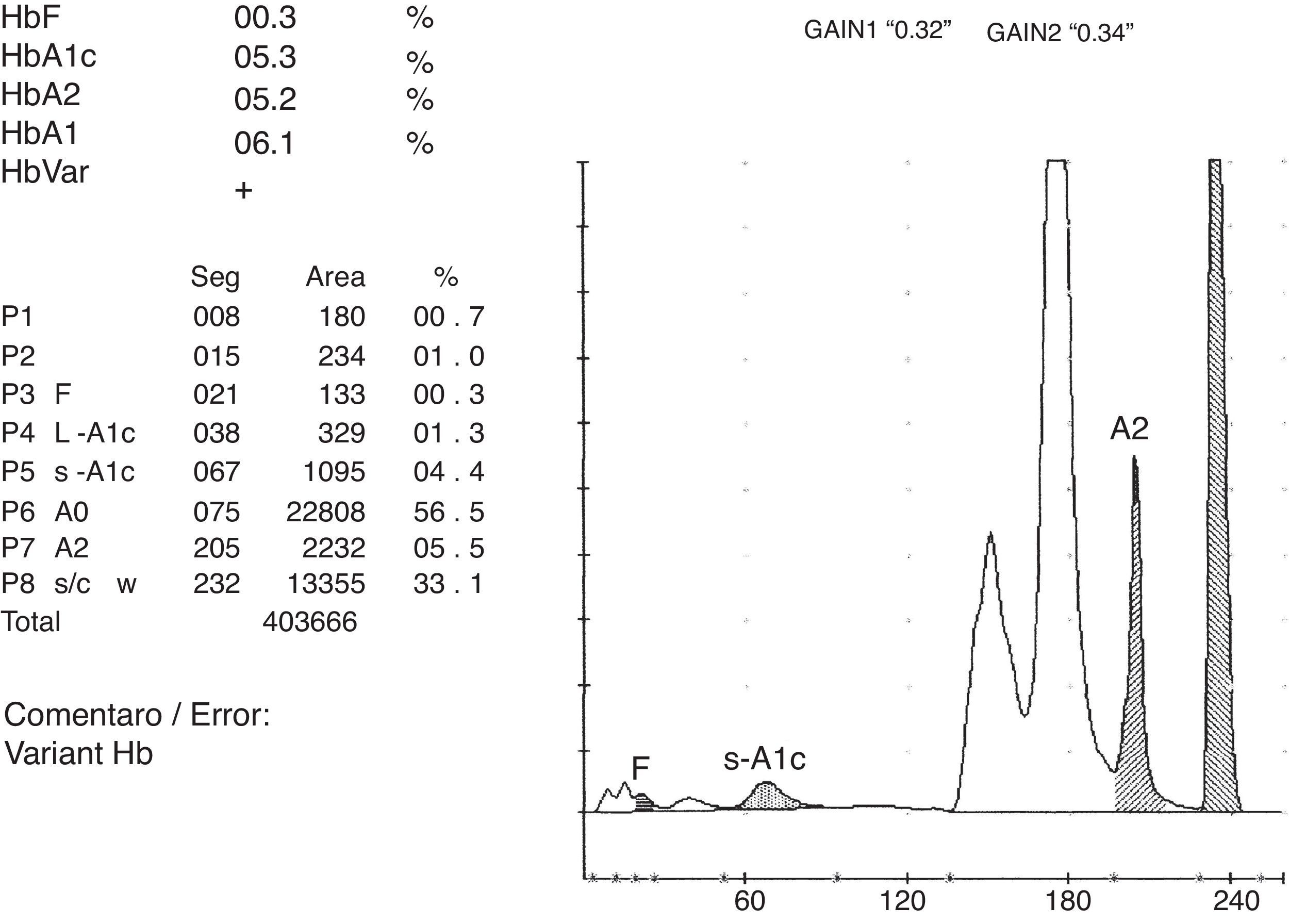
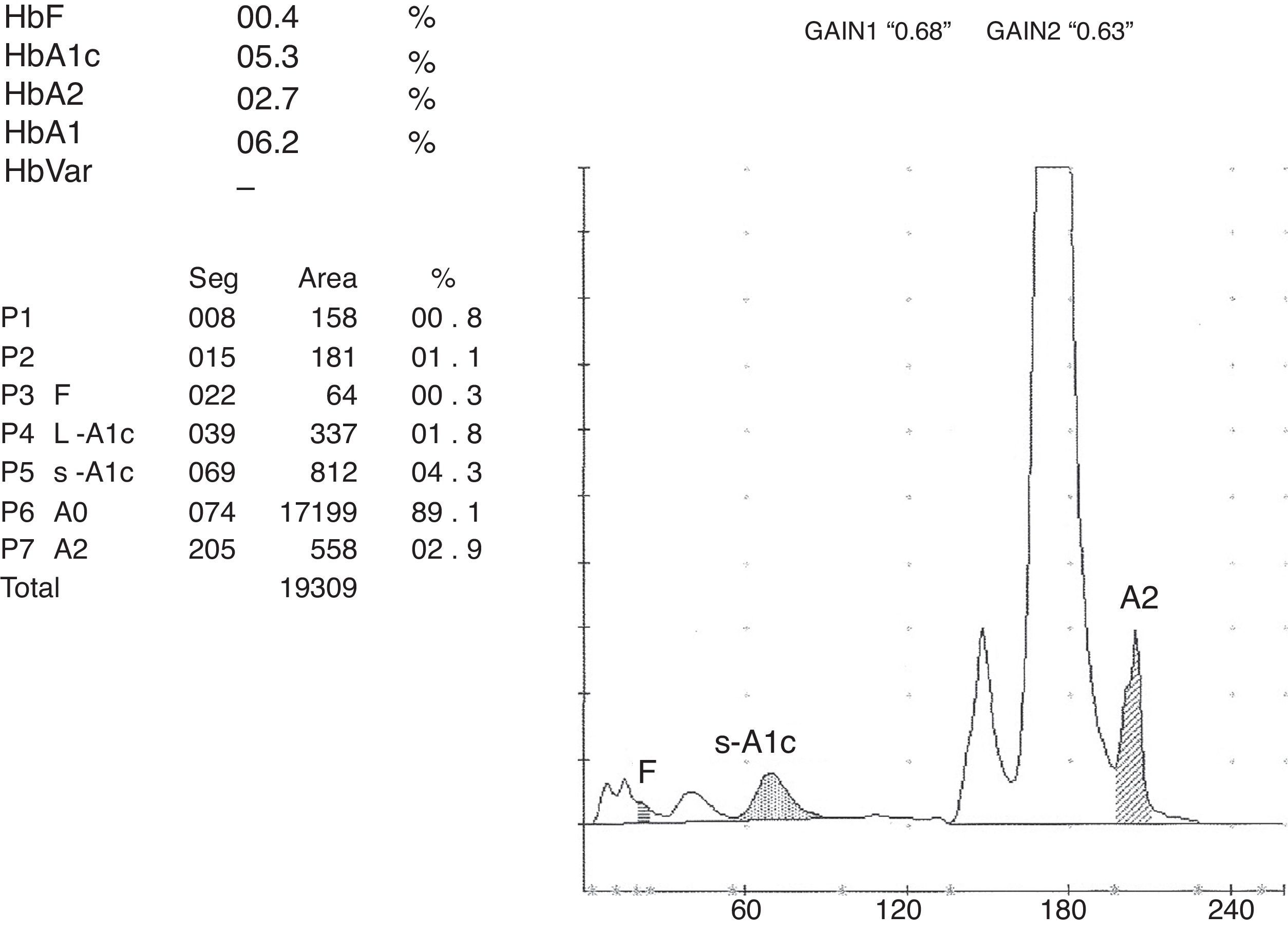
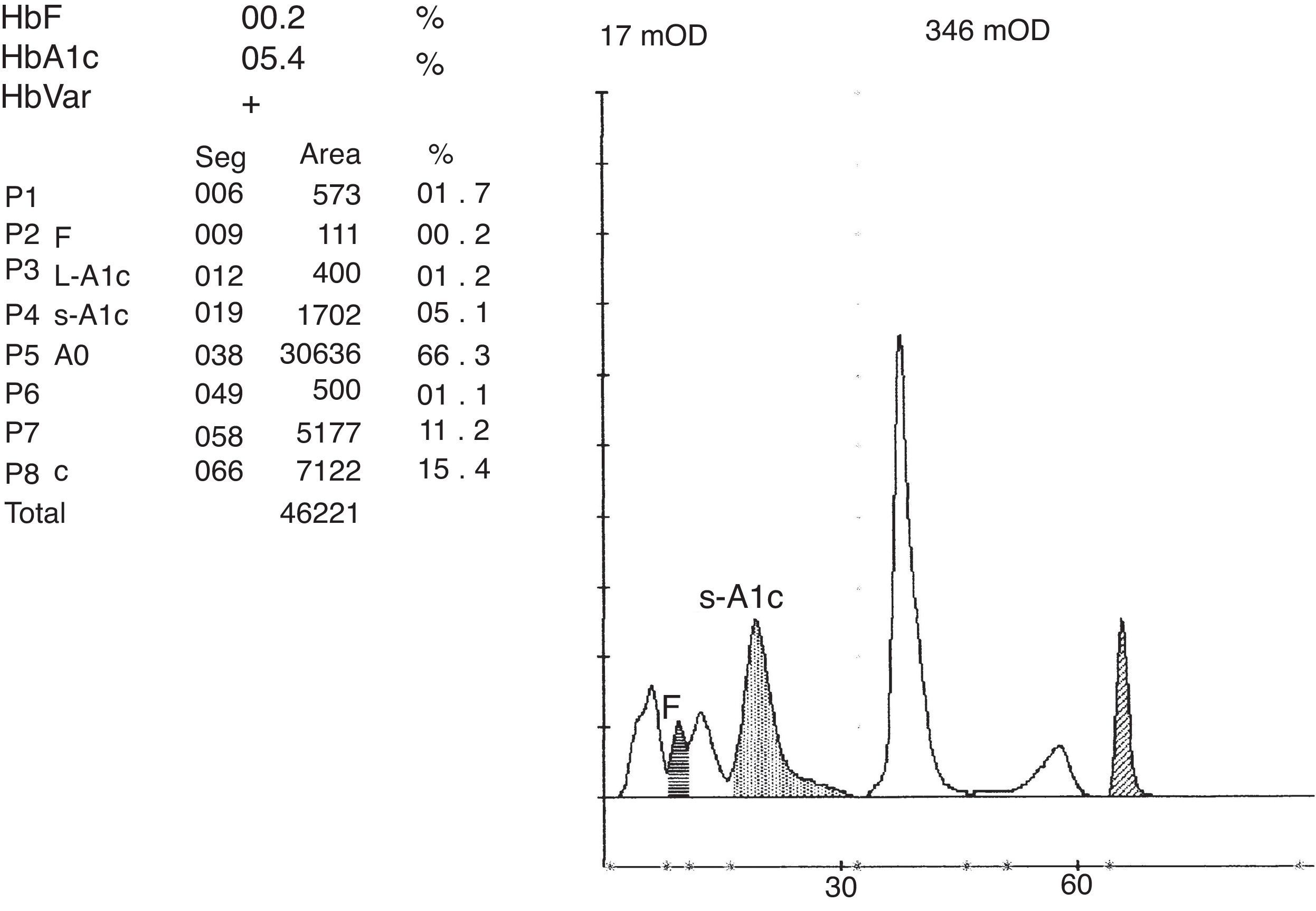
ResultadosEn el hemograma destacó Hb13,9g/dl, hematíes 6,03×106/μl, hematocrito 38,7%, VCM 64,3fL, CHM 23,1pg, CHCM 36g/dl y ADE 14%. Metabolismo férrico normal: hierro 101μg/dl (normal 60-180), ferritina 35 ng/ml (10-120), transferrina 321mg/dl (200-360), índice de saturación de transferrina 22% (15-30). Sin datos de hemólisis: bilirrubina total 0,7mg/dl (0,3-1,2), LDH 213 UI/l (0-247), haptoglobina 134mg/dl (50-320), reticulocitos manuales 1,2%. Al detectar microcitosis e hipocromía sin datos de ferropenia se inició estudio de posible talasemia. En el frotis sanguíneo únicamente se observó una discreta microcitosis e hipocromía, sin otros hallazgos morfológicos. Al realizar la determinación de HbA2 y F por HPLC se detectó una variante anómala por el sistema ADAMSTM A1c HA-8160 en el modo talasemia, que comporta un 33,1% (figs. 1 y 2). Por el sistema ADAMSTM A1c HA-8180V de mayor resolución se detectó una banda anómala que se situó en la zona de la HbC, y que cuantificó como un 15,4% (fig. 3), lo que justificaría la hipocromía; HbA25,2% y F0,3%. Se solicitó estudio genético al centro de referencia de las cadenas α y β, realizando técnicas de PCR multiplex, y concluyendo en una sola deleción 3,7Kb en la cadena α y patrón normal en el estudio de las 22 mutaciones disponibles de la cadena β (incluyendo la correspondiente a la HbC). Ante los resultados discordantes en el estudio de la cadena β se envió muestra a otro centro más especializado, que procedió a la amplificación por PCR y secuenciación del fragmento amplificado, identificando la mutación β 121 (Glu>Lys), en estado heterocigoto, compatible con HbO-Arab.
 en el modo talasemia cuantificada en la ventana S/C, pico 8, como un 33,1%.")
.")
 detecta una banda anómala que se sitúa en la zona de la Hb C y que cuantifica como un 15,4%, pico 8 (Hb O-Arab). Adviértase el pico presente antes de la banda anómala.")
En la provincia de Almería, y según datos del INE 2013, la población inmigrante supone el 20,8% del total (144.693 de 696.159 habitantes), la mayoría con origen en el Magreb y África subsahariana. Debido a estos movimientos migratorios estamos asistiendo en nuestra área a enfermedad hasta ahora muy poco frecuente en nuestro medio, como es el caso de la HbO-Arab, una variante poco habitual de hemoglobina debida a una mutación puntual de la cadena β, descrita en afroamericanos, países balcánicos, árabes y egipcios.
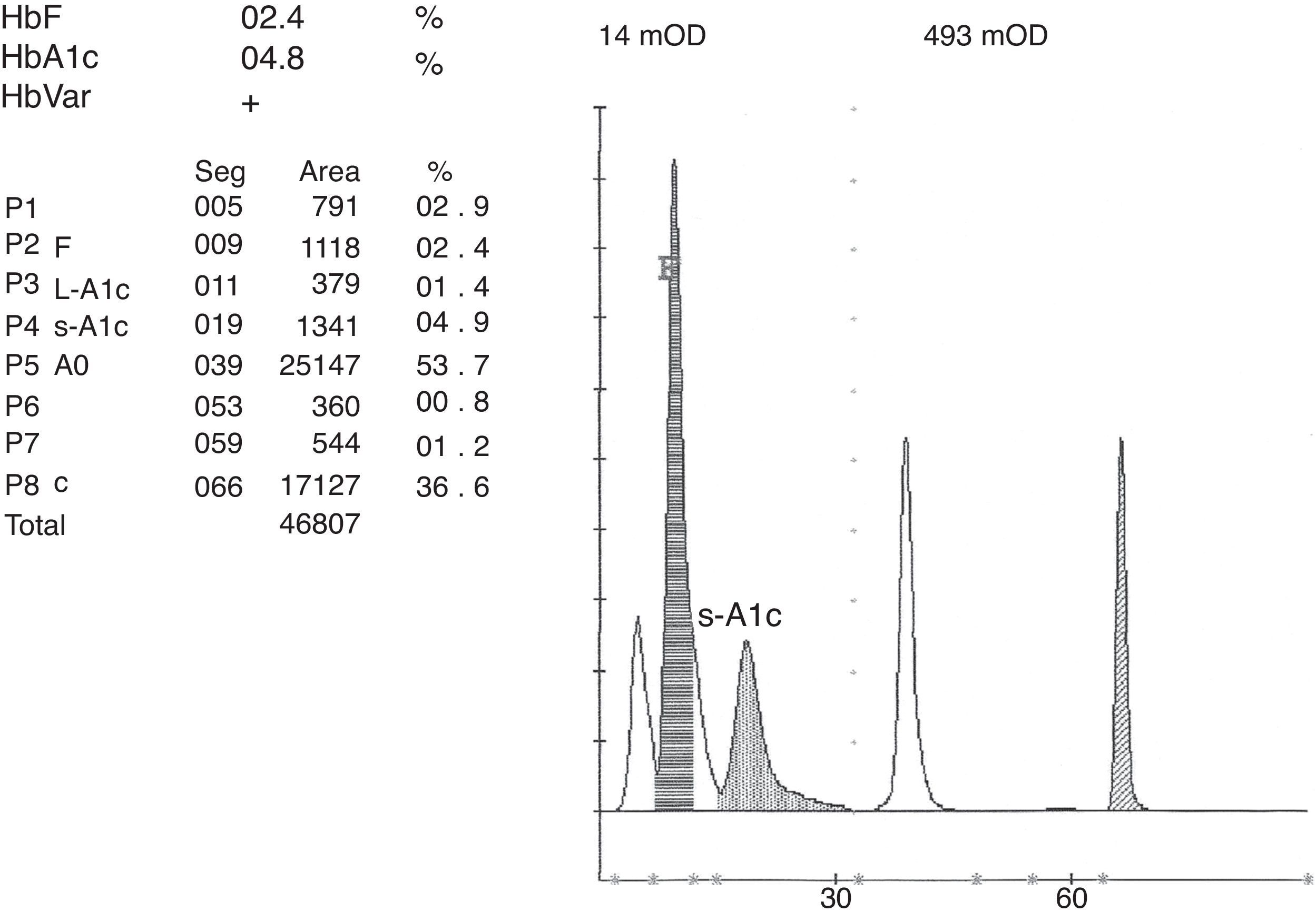
Los sistemas HPLC se encuentran disponibles en todos los laboratorios, puesto que se utilizan de forma sistemática para el diagnóstico y control de la diabetes mellitus con la medición de la HbA1c. Desde que en 1983 se adaptaron al diagnóstico de hemoglobinopatías5, estos sistemas ofrecen un método rápido, simple, asequible y fiable, altamente eficaz en la detección de hemoglobinopatías. Se han descrito un número significativo de dichas variantes que interfieren en la cuantificación de la HbA1c6. Con los equipos ADAMSTM A1c HA-8160 y HA-8180 (A. Menarini Diagnostics®) las variantes de HbS y C, o aquellas que se movilizan a sus zonas, como en nuestro caso la O-Arab en la zona de la HbC, no afectan a la medición de HbA1c y son explícitamente identificadas y correctamente cuantificadas7. Se ha descrito que con el sistema Bio-Rad Variant II® es posible distinguir la HbO-Arab de la HbC por la presencia de un pico previo a la banda anómala8. Con el sistema ADAMSTM A1c HA-8180 igualmente se puede observar un pico anterior a la banda de la HbO-Arab (figs. 3 y 4), lo que también haría posible la distinción entre ambas hemoglobinas.
 detecta una banda anómala identificada como Hb C, cuantificada como un 36,6%. Adviértase la ausencia del pico antes de la banda, como ocurre con la Hb O-Arab de la figura 3.")
Una característica importante es que, al contrario que los métodos de electroforesis en gel, los sistemas HPLC ofrecen una cuantificación precisa de la cantidad de HbA2 y F, datos que proporcionan el diagnóstico de β y δβ talasemias. Por otro lado, la movilidad de las variantes estructurales de Hb expresa un tiempo de retención constante y reproducible, facilitando su interpretación. Junto con los índices hematimétricos y un test de falciformación se puede caracterizar la hemoglobinopatía más frecuente y de mayor repercusión clínica, como es la HbS. Sin embargo, la identificación definitiva de las variantes anómalas requiere de técnicas moleculares3.
Si bien la asociación de HbO-Arab con HbS es causante de una forma grave de enfermedad drepanocítica, la asociación con α-talasemia heterocigota conlleva únicamente la aparición de microcitosis. La α-talasemia es muy frecuente en el área mediterránea y en la población africana. Dado que las hemoglobinopatías estructurales más frecuentes (HbS, HbC, HbE, HbD, HbO-Arab, defectos de la cadena β) y la α-talasemia se localizan en cromosomas distintos, se heredan de forma independiente. Cuando ambos defectos se asocian en una doble heterocigosidad se dispone de un menor número de cadenas α para unirse a las cadenas β normal y anómala; en estos casos las cadenas α que quedan se unirán preferentemente a la cadena β normal, lo que reduce el porcentaje de Hb anómala. De ahí que, como en nuestro caso, la asociación de α-talasemia puede llevar a un nivel anormalmente bajo de la Hb variante, lo que puede sospecharse si existe microcitosis. Por otro lado, la α-talasemia no muestra características específicas en la HPLC.
Como en nuestro caso, la elevación de HbA2 por encima del 3,5% por HPLC se considera falseada en presencia de variantes de hemoglobina3,9. En este sentido es importante tener en cuenta que el hallazgo de microcitosis no debe conllevar un diagnóstico erróneo de β-talasemia, sino que debe investigarse la asociación con α-talasemia, más aún cuando existe un nivel anormalmente bajo de la variante de hemoglobina, como se ha descrito previamente.
En resumen, hoy en día con los movimientos migratorios estamos atendiendo a población portadora de hemoglobinopatías hasta hace unos años muy infrecuentes en nuestro medio. Es por ello que nos vemos obligados a establecer estrategias diagnósticas integradas y multidisciplinares para su estudio10, con la implicación de distintas áreas diagnósticas y clínicas, y que incluyan la interpretación de los cromatogramas ofrecidos por HPLC, la valoración de los índices hematimétricos y la evaluación clínica del paciente en cuanto a origen, etnia y sintomatología.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Servicio de Genética del Hospital Clínico de Granada (en especial a Dolores Maciá Trives) y a la Unidad de Anemias y Eritropatología y Unidad de Genética Molecular Hematológica del Hospital Vall d’Hebron, a través de Menarini Diagnostics S.A., por su aportación en los estudios moleculares.
