


Los linfomas de glándula parótida son una entidad poco frecuente: se estima una incidencia del 0,3% de todos los tumores malignos del organismo y de un 4% de todos los linfomas1,2. Dentro de los linfomas parotídeos, el 80% corresponde a linfomas no Hodgkin, y son los tipos folicular, difuso de células grandes y los asociados a mucosas (MALT) los más frecuentes.
En torno al 30% de los linfomas MALT afectan a cabeza y cuello. Por orden de frecuencia se sitúan en el anillo de Waldeyer, senos paranasales y glándulas salivales (parótida 75%; submaxilar 25%), pero también se describen en otras áreas como globo ocular, tiroides, timo, pulmón, etc.1,3.
El propósito de este artículo es presentar un caso clínico infrecuente de lesión parotídea de difícil diagnóstico y su manejo clínico posterior.
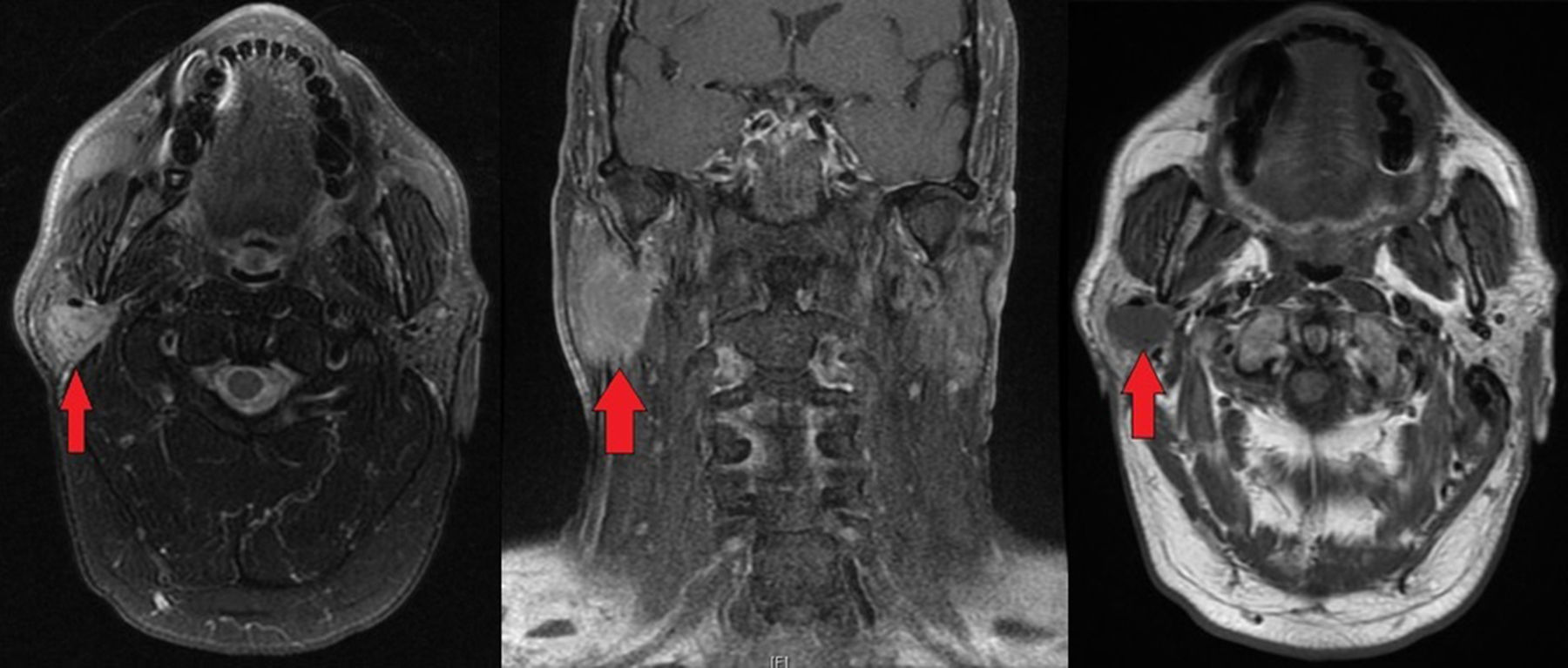
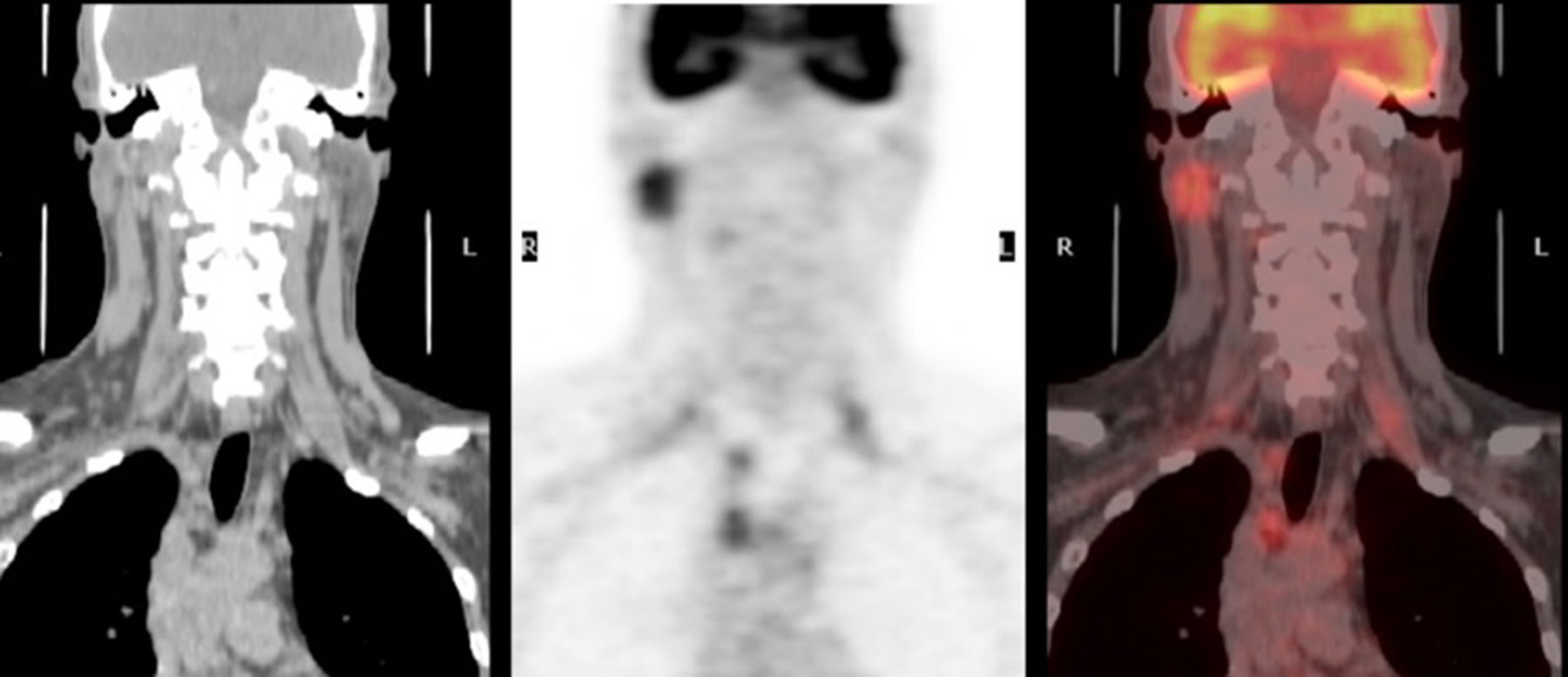
Caso clínicoPresentamos el caso de un varón, de 55 años, sin antecedentes de interés, que acudió a consultas por tumoración parotídea derecha de un mes de evolución. A la exploración se observó House-Brackmann I bilateral con tumoración pétrea, no dolorosa y fija a lóbulo profundo parotídeo de aproximadamente 2cm de diámetro, sin lesiones en otros niveles. Se practicó estudio mediante ECO-PAAF que informó una lesión de 17×14mm en el lóbulo superficial y profundo de la parótida derecha con resultado anatomopatológico de linfoadenitis reactiva. La RMN precisó una lesión de 18×24mm en el lóbulo profundo y superficial con adenopatías asociadas supraclaviculares y paratraqueales ipsolaterales sugestivas de malignidad (fig. 1). Ante estos hallazgos, se realizó PET-TAC que informó de múltiples focos sugestivos de malignidad: lesión nodular sólida parótida derecha con SUV 6, adenopatías supraclaviculares, retrotraqueales y paratraqueales ipsolaterales con SUV 6,4 (fig. 2).


Ante la sospecha de una tumoración maligna parotídea, nos planteamos como posibilidades diagnósticas la presencia de un carcinoma mucoepidermoide de alto grado, un adenocarcinoma o, con mayor probabilidad debido a la negatividad de la PAAF, un carcinoma sobre adenoma pleomorfo o un tumor con estirpe linfoide como los linfomas o carcinomas linfoepiteliales, los cuales suelen presentar metástasis ganglionares cervicales al momento del diagnóstico en más de un 40% de los casos.
Por todo ello, se decidió tratamiento quirúrgico para diagnóstico intraoperatorio y valorar cirugía (parotidectomía y vaciamiento cervical), según los hallazgos.
Al mes desde la primera consulta, se intervino al paciente mediante parotidectomía superficial, con hallazgo anatomopatológico intraoperatorio de proceso linfoproliferativo benigno que infiltraba la parótida derecha, por lo que no se realizó parotidectomía total ni vaciamiento ganglionar.
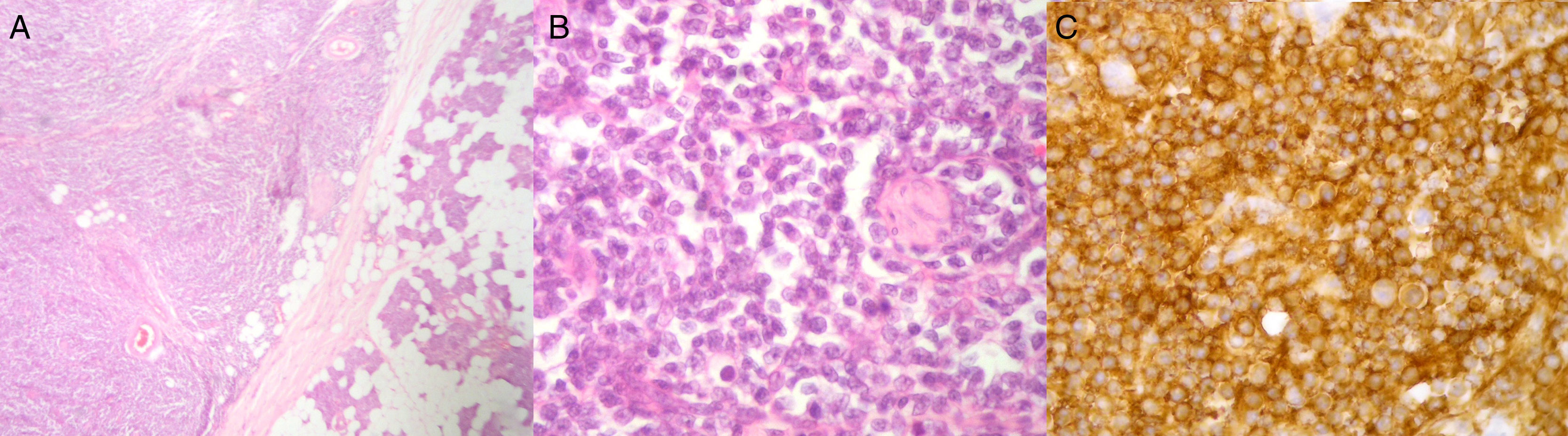
El diagnóstico anatomopatológico definitivo con empleo de pruebas de inmunohistoquímica informó de linfoma MALT de la zona marginal (fig. 3). El paciente fue derivado al Servicio de Hematología, donde completó tratamiento mediante esquema R-CHOP (rituximab, ciclofosfamida, doxorrubicina, vincristina) durante 4 meses. El control posterior en consultas y las pruebas de control mediante TAC y PET-TAC mostraron una completa resolución del proceso al finalizar el tratamiento, sin evidenciar recidivas en los 3 años de seguimiento.
 Infiltración de la parótida por una proliferación difusa de tipo linfoide (hematoxilina-eosina, 40x). B) Detalle de la proliferación constituida por linfocitos de pequeño tamaño, núcleo redondo y escaso citoplasma (hematoxilina-eosina, 400x). C) Positividad inmunohistoquímica intensa y difusa para CD20 (400x).")
A) Infiltración de la parótida por una proliferación difusa de tipo linfoide (hematoxilina-eosina, 40x). B) Detalle de la proliferación constituida por linfocitos de pequeño tamaño, núcleo redondo y escaso citoplasma (hematoxilina-eosina, 400x). C) Positividad inmunohistoquímica intensa y difusa para CD20 (400x).
El linfoma MALT es un subtipo de linfoma de células B extranodal de bajo grado descrito por primera vez por Isaacson y Wright en 19834 que afecta principalmente al tracto gastrointestinal (70%), pero que también se puede presentar en gran variedad de localizaciones (glándulas salivales, globo ocular, tiroides, timo, pulmón, mama, riñón, hígado, próstata). Las glándulas salivales no contienen normalmente tejido MALT, pero este puede ser adquirido como resultado de una inflamación crónica1,5. Hasta en el 44% de estos pacientes se asocian enfermedades autoinmunes (lupus eritematoso sistémico, tiroiditis de Hashimoto): el síndrome de Sjögren es la entidad más común, que incrementa 40 veces el riesgo de linfomas parotídeos (80% linfomas MALT)2,6. También se ha descrito, en esta inflamación crónica susceptible de transformación a linfoma MALT, la participación de agentes infecciosos como VEB, VHC, VIH, Helicobacter pylori, Borrelia o Clamidia psitacii7.
Los linfomas MALT son más frecuentes en la 6.ª década de la vida, con predominio en mujeres sobre varones (3/1), en probable relación con la mayor afectación de enfermedades autoinmunes. Presentan un curso indolente con diseminación lenta; al diagnóstico, el 50% de los casos presentan afectación multifocal sincrónica. En algunos casos se pueden transformar en linfomas difusos de células grandes5,8,9.
El diagnóstico de los linfomas MALT parotídeos es similar al resto de los tumores de esta área. Se emplean TAC y RMN con hallazgos de lesiones quísticas inespecíficas que pueden asociar calcificaciones. El uso de la PAAF en linfomas de cabeza y cuello presenta una tasa del 32% de falsos negativos9, por lo que nunca podremos descartar su sospecha ante resultados inespecíficos. En cuanto al papel del PET en los linfomas, su precisión depende del subtipo y de la localización: es más fiable para los linfomas de localización nodal y menos para los extranodales, y detecta el 67% de los linfomas MALT de la zona marginal1. Constituye una prueba fundamental tanto en el diagnóstico inicial para detectar formas multifocales como en el seguimiento posterior tras tratamiento.
El tratamiento de linfomas MALT es controvertido. Se recomienda quimioterapia con o sin radioterapia asociada1,6 en las formas diseminadas o tras la resección incompleta del tumor, mientras que la cirugía exclusiva (parotidectomía) queda reservada para tumores de localización única, aunque existen autores que describen mayor riesgo de diseminación solo con tratamiento local y recomiendan asociar siempre quimioterapia o radioterapia10.
El pronóstico es excelente, con una supervivencia mayor al 80% a los 5 años y superior al 75% a los 10 años, cifras mejores que en el resto de los linfomas de cabeza y cuello1.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
