


Craniosynostosis refers to a great number of deformities associated with the premature fusion of cranial sutures. There are several types of synostoses, depending on the number of sutures affected and the involved regions. Surgery is indicated for functional and esthetic reasons.
Although many surgical techniques have been described, the decision of the type of operation to be performed and the time of the procedure should be based on three pillars: the patient's clinical and tomographic features as well as social and demographic aspects; the expertise of the treating surgeons; and the resources available at the craniofacial center. The patient's outcome will be directly related to a structured management protocol, implemented by a multidisciplinary team, in which all team members should speak the same language.
The main objective of this article is to describe the management protocol developed at a public tertiary level medical institution in Mexico.
El término craneosinostosis hace referencia a una gran variedad de deformidades asociadas a la fusión prematura de suturas craneales. Existen varios tipos de sinostosis, de acuerdo al número de suturas y regiones involucradas. El manejo quirúrgico está indicado por razones funcionales y estéticas.
Numerosas técnicas quirúrgicas han sido descritas. La decisión en cuanto a la técnica quirúrgica a realizar y el tiempo de la intervención deberá basarse en tres pilares: los hallazgos clínicos y tomográficos del paciente, así como los aspectos socio-demográficos relacionados con el mismo; la experiencia de los cirujanos tratantes; y los recursos disponibles del centro craneofacial. El resultado obtenido estará asociado directamente a un protocolo de manejo organizado, llevado a cabo por el equipo multidisciplinario involucrado, en el cual todos los miembros deberán hablar un mismo lenguaje.
El objetivo principal de este artículo es describir el protocolo de manejo en una institución médica de tercer nivel en México.
Craniosynostosis is defined as a premature fusion of the cranial sutures, and has an incidence of 1/1750 to 1/2100 live births.1–4 There is a wide array of clinical features, from isolated suture craniosynostosis, to multiple cranial sutures being fused (complex craniosynostosis), or syndromic craniosynostosis, where patients show abnormalities involving the skull and other regions of their bodies.5 According to the classification proposed by Thompson and Hayward,6 single-sutural synostoses (isolated), complex craniosynostosis and syndromic craniosynostosis, pertain to the primary craniosynostosis group. Secondary craniosynostosis refers to premature fusion of skull sutures, which happen in association with metabolic abnormalities (e.g. hypophosphatemic rickets, hyperthyroidism), hematologic conditions (e.g. polycythemia vera or thalassemia), and abnormalities related to mucopolysaccharides storage (Hurler and Morquio syndromes), among others.6
Although several theories to try to explain its causation have been postulated, the main process by which this abnormal fusion occurs remains unclear.7 Nevertheless, craniosynostosis has been linked to numerous factors: genetic, metabolic and demographic.8–15 Valproic acid has been associated with trigonocephaly.8,9,16 Antacids used in infants with abdominal colic have also shown a relation.12 Syndromic synostoses are associated with genetic mutations, specifically with genes thought to be involved in the regulation of osteogenic lineage, such as the TWIST and fibroblastic growth factor receptor (FGFR).17–19 The syndromes more frequently found to display a genetic component are the following: Crouzon (chromosome 10q), Saethre-Chotzen (chromosome 7p) and Pfeiffer (chromosome 8p).20 Most of the syndromic craniosynostoses display an autosomal dominant inheritance.17,18 It is much more uncommon for complex and isolated suture craniosynostosis to be associated with these genetic mutations.
Incidence of isolated suture craniosynostosis is about 1 in 2000 live births and the gender more frequently affected is the male gender.1–3 The sagittal suture is the more frequently involved suture (40–60%).4,5,21 The observed trend for the metopic suture becoming the second most common type has been seen at our institution. This is followed by the unilateral coronal suture. The objectives of the following article are to describe the study and management protocol of patients with craniosynostosis at a public, third level institution, taking into account the social and demographic factors of the patient, in a developing country.
Diagnosis and preoperative evaluationThe patient with possible craniosynostosis must have a multidisciplinary approach from the beginning. Once the pediatrician has the suspicion of a fused cranial suture, the patient must be referred to the craniofacial clinic, where the craniofacial surgeon and the pediatric neurosurgeon will be involved with the assessment.
Clinical diagnosis can be established in several isolated suture craniosynostoses, specifically scaphocephaly and trigonocephaly. The computed tomography with tridimensional craniofacial reconstruction will help confirm diagnosis in less common craniosynostosis (e.g. lambdoid) or in multiple-suture synostoses, and will be a valuable aid in planning surgery. In addition, tomographic information might be extremely useful to determine the location and characteristics of intracranial sinuses and other associated conditions to specific types of craniosynostosis, such as a “pseudo” nasal encephalocele, where the brain extends between the orbits, in metopic suture synostoses.22
Regelsberger et al.23 confirmed using ultrasonography the diagnosis of craniosynostosis, where bony bridges showed as hyperechogenic structures with and without depressions. Although it represents a non-invasive procedure, without radiation exposure, and less expensive method, ultrasonography is less reliable in patients older than 12 months of age, due to increased bone thickness and reduced widening of the intersutural space. Methods, such as the plagiocephalometry, in which a strip of thermoplastic material is placed on the head's maximal transverse circumference,24 may be another non-invasive, clinically based procedure; nevertheless, it has the disadvantage of the learning curve for the medical personnel who performs it and the lack of information regarding the skull's height.25
When the patient has suggestive symptoms of intracranial hypertension, a study to determine an exact quantitative level of the intracranial pressure might provide more information than the mean intracranial pressure.26 The transcranial Doppler ultrasound is a non-invasive method to establish the intracranial pressure, based on indexes of mean pulsatility and resistance over the sagittal sinus.27
Surgical indications: which patients must undergo a cranioplasty procedure?Surgery is required in patients with craniosynostosis to treat the negative effects of this condition on the brain development, due to impaired regional cerebral blood flow secondary to an increased intracranial pressure, in areas of restricted skull growth, and to correct the abnormal cranial shape due to altered patterns of growth.5 There are selected cases of synostoses in which no surgical treatment is required. Whether partial sutural closure with minimal changes in skull shape requires surgical treatment is still a debatable topic. It is logical to think that if no clinical symptoms or cognitive impact can be demonstrated, these patients can be followed conservatively. On the other hand, a dilemma occurs in patients with multiple suture synostosis, in whom an apparent normal head shape is present and intracranial pressure shows upper limit values with subtle clinical symptoms.5,28 Patients who present with craniosynostosis and an apparent normal head shape, with associated delayed milestones and in whom it is difficult to assess the cognitive and psychomotor development, due to lack of clinical records secondary to poor family or social support, or patients who live in distant, rural areas with an unsatisfactory clinical surveillance, it becomes even more decisive for the individualized, careful analysis by the multidisciplinary team members.
The time of the operation has been a constant topic of discussion in craniofacial forums. The advantages of performing corrective surgery before 8 months of age are as follows: increased osseous healing; softer, malleable bone, more amenable to external devices to reshape the head; a decreased psychological impact for parents and a decreased surgical time with associated decreased blood loss. Arguments in favor of surgery after 8 months of age are as follows: thicker bone with more stable fixation, an increased circulating blood volume and more mature organs for a patient undergoing a major anesthetic and surgical event. Some authors suggest that surgery performed after 10 months of age helps to take advantage of the rapid skull growth during the first year of age.29 Patel et al.30 document better neuropsychological outcomes on patients with sagittal craniosynostosis who underwent surgery before 6 months of age compared with those who had a procedure performed between 6 and 12 months of age. Regardless of the time of operation, surgery should be performed before 1 year of age, as studies document a decreased intellectual development in patients with bicoronal synostosis who underwent a cranioplasty after 12 months of age, compared with patients with the same condition operated before their first anniversary.
In Mexico, social, cultural, demographic and economic factors play a determinant role in the treatment of craniosynostosis. Oftentimes, patients are referred rather late (around 1 year of age) for the first evaluation by the craniofacial team. This is partly due to an insufficient medical surveillance and follow-up in patients and families who live in rural areas and small towns, and in families with a poor cultural background. Having said that, these patients are only candidates for extensive remodeling cranioplasty procedures.
Surgical options: what choice is suitable for the patient?Overall, surgical techniques to correct craniosynostosis can be classified in two groups: procedures to remove the involved suture (strip craniectomy) and techniques that remodel the skull (cranioplasty).5 The former group can be subdivided in procedures that remove the fused suture and procedures that extract the involved suture and use devices to modify the skull shape postoperatively.5 Among the devices used are the following: restrictive/molding external helmets, and springs or osteogenic distractor device. Jimenez and Barone31 described a hybrid technique in which patients undergo an endoscopic strip craniectomy followed by postoperative molding helmet, with the objective of achieving a more normal head shape than when the involved suture is resected only. Other authors have documented that this hybrid technique is associated with reduced transfusion rates (7–26%)32–34 and shorter stays in hospital.30,35,36 With regard to the use of osteogenic distraction devices and springs, the fact that the expansion rate is difficult to control and that the patient will require at least two surgical events, raise the question of its real benefits. In addition, complication rates are similar to other techniques.37,38
Cranioplasty techniques confer several advantages as follows: to treat and remodel, in a controlled fashion, multiple skull regions at the same surgical event, the constricted areas can be expanded, and the areas that show an increased diameter due to compensatory overgrowth can be reduced.39 Other benefits include more normal cranial indexes, when compared to strip craniectomy procedures.40 The drawbacks include the following: a more extensive surgical procedure with greater technical difficulty, increased blood loss and increased transfusion rates, with its potential adverse effects.41,42
Intraoperative blood loss represents one of the commonest causes of death in patients with craniosynostosis submitted to surgical correction.43 Several adjuvants to reduce the blood loss, such as recombinant erythropoietin, administered preoperatively, have demonstrated reduction in blood product transfusion significantly.44 A positive impact in decreasing transfusion of blood products in patients with craniosynostosis has also been found with the use of tranexamic acid.45 Use of cell saving devices represents an excellent option for patients operated on their cranial vaults, especially when one or more blood volumes are lost.46–48 Nevertheless, the latter option requires especial devices, and products, as well as trained personnel, which adds a significant expense in treatment. In addition, there is a reduced availability of these products and devices in public medical institutions in Mexico and Latin America. Therefore, in these medical settings, other less effective mechanisms to try and reduce transfusion of blood products, such as hypotensive anesthesia and hemodilution, must be performed.
Cranioplasty procedures involve removing the affected bones (synostotic and non-synostotic regions), remodeling them by the use of techniques that cut and bend bone on specific sites and repositioning the manipulated bone segments in specific areas. In order for an adequate volume and shape to be achieved, the bone segments must be fixed in a stable position. Numerous materials to pursue this goal are available, and can be basically classified in non-absorbable (e.g. stainless steel wire, titanium alloy plates and screws) and absorbable (sutures and lactic acid plates and screws). Although extensively used in the last few decades of the twentieth century, non-absorbable materials are no longer recommended in pediatric patients due to a potential intracranial migration with growth. On the other hand, absorbable plates and screws are planned to confer stability during the period where bony consolidation and healing must take place. Nevertheless, these materials might be present for long periods of time after surgery, and might be associated with sterile collections or draining abscesses.49–52 Furthermore, these products add a substantial cost to the overall treatment. Resorbable sutures, such as PDS II [Ethicon, USA], may well be an excellent option to provide a stable fixation, although more technically advanced fixation techniques5 and longer operating times are required.
In selecting the surgical technique to be performed, the multidisciplinary team must take into account the social and demographic factors presented individually by each patient and the technical expertise, and the available medical and non-medical resources, and the potential pitfalls in the specific medical setting. For example, a patient from a rural area with a poor family and social support will not be a good candidate for a strip craniectomy and the use of external devices, as the follow-up period will not be as consistent. Therefore, a more extensive procedure must be used. On the other hand, a malnourished baby, a so common problem in developing and under-developed countries, will have an increased risk in spite of multiple efforts to correct the nutritional deficiency. Thus, a less invasive procedure might represent his best option.
It must also be kept in mind that in some patients, the affected cranial regions, may show a behavior toward recurrence of the initial pathology, which may condition persistent abnormalities that need to be addressed with subsequent surgical procedures.
Complex and syndromic craniosynostosis: changes in the treatment algorithmComplex craniosynostosis represents a much more surgically demanding problem from the technical point of view, as oftentimes the abnormalities shown by the patient are not only the sum of the fused sutures, but the compensatory effects on other skull regions, which become indirectly affected as well. Furthermore, the management protocol may be difficult due to an associated Chiari malformation (a caudal displacement of cerebellar tonsillar structures through the foramen magnum). Patients with syndromic craniosynostosis, on the other hand, in addition to multiple sutures being fused, may demonstrate involvement of other areas such as the face and hands. For example, in Apert syndrome, a midfacial retrusion occurs, with a potential negative impact on breathing, which may translate in obstructive sleep apnea; features in addition to hands and feet abnormalities, such as syndactyly.1,53 Abnormal airways add a significant increased difficulty for the pediatric anesthetist and the postoperative medical and nursing personnel, as well as require several polysomnography and related studies to follow ventilatory disturbances.1,54
Since treatment of these patients may involve multiple procedures and, thus, be affected by numerous potential complications, a liaison must be established between the affected family and the multidisciplinary team. It is imperative to discuss with parents or guardians that treatment will be a process, rather than a single operation. Social and demographic factors play a crucial role in developing countries, as oftentimes families of affected children, have a humble cultural and educational background which has a direct impact on the postoperative care due to poor support, or deficient responsibility, for these patients. With that in mind, it is essential to gather as many tools and resources in order to strengthen the familiar and social network, in which the multidisciplinary team explains clearly and in lay-language the characteristics and potential pitfalls of each and every step of the treatment protocol. Again, constant supervision of the patient and parents/guardian will help avoid hurdles along this complex road.
Preoperative and postoperative management protocolThe preoperative and postoperative management must take into account two classes of patient's aspects: the clinical and the socio-demographic. The former can be subdivided into the following: the nutritional status, overall health and comorbidities, airway characteristics, in addition to achieved milestones, cognitive deficits, conduct and behavior, and signs and symptoms of intracranial hypertension. The latter most consider the geographic location of the patient's home and the available resources at that specific region, the distance from the treating medical facility and available transport, and the type and proximity of the closest medical practitioner/pediatrician who follows the patient. It is of great importance to clearly identify the parent/guardian who will be taking care of the patient and to determine the overall family and social support. Lay language must be used at all times, aiming to consolidate a patient–craniofacial center liaison.
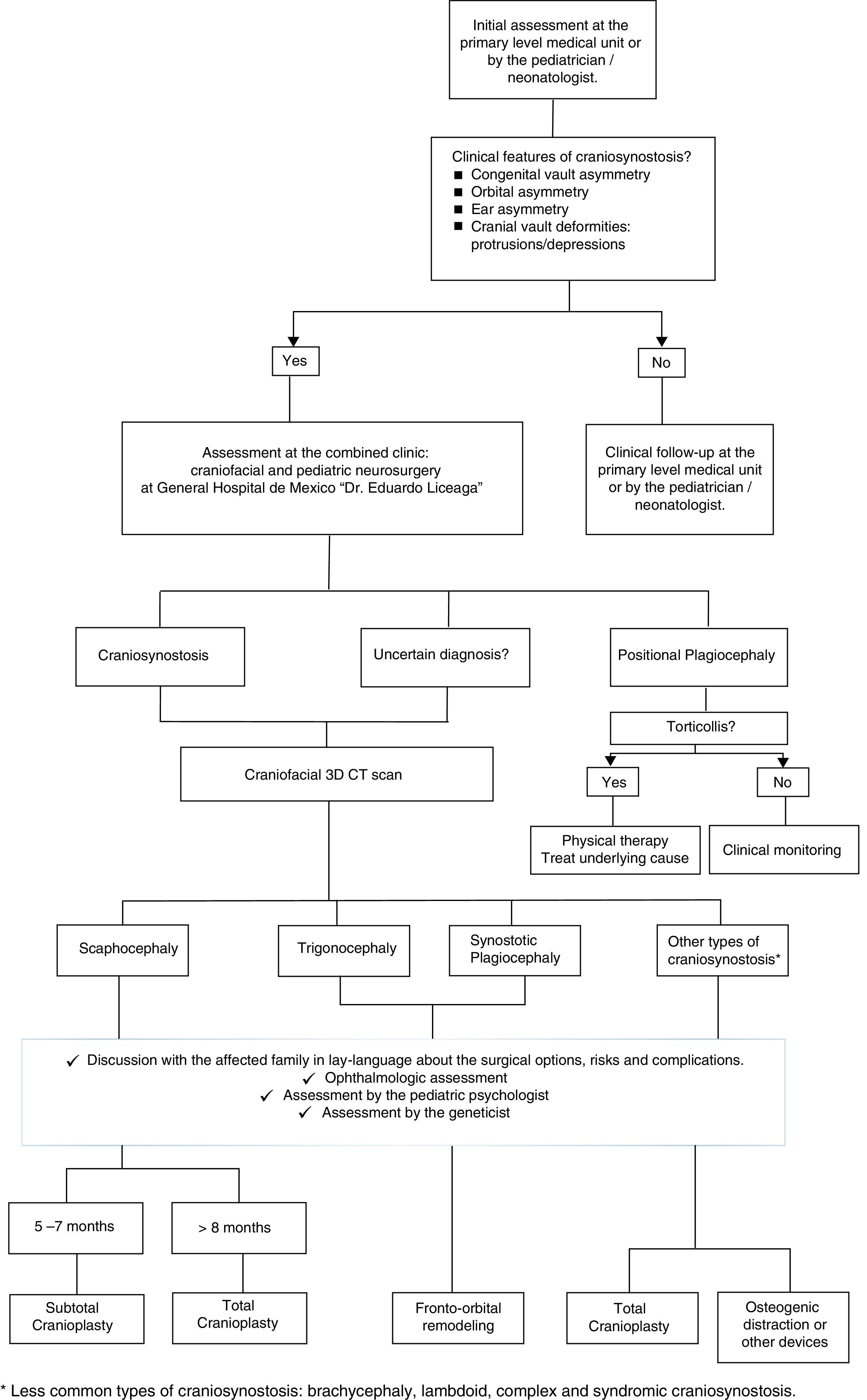
As shown in Fig. 1, the patient suspected to present clinical features of craniosynostosis (e.g. congenital head asymmetry, orbital asymmetry, ear asymmetry or prominent areas or depressions on the skull) by the pediatrician/neonatologist must be referred as early as possible to the craniofacial center. If these features are confirmed or if the diagnosis is uncertain, a craniofacial tridimensional CT scan must be performed. This exam will aid in determining the type of synostosis. With the appropriate information, the family must be explained in lay terms about the possible surgical options, risks and complications; always taking into account the social and demographic factors previously mentioned. In addition, the baby must be examined by the ophthalmologist, the pediatric psychologist and by the genetics department. Based on the type of craniosynostosis and clinical features, the patient's age and social and demographic factors; and the technical expertise of the practicing surgeons, the surgical technique will be selected.

In craniofacial centers where resources are limited, planning takes an even more important role, where potential pitfalls and complications must be extensively discussed by all team members previous to the operation. In addition to a central and an arterial line, blood transfusion products must be available and planned based on the patient's estimated blood volume and accurate blood loss monitoring. The room temperature should be kept warm, and if this is difficult, a warming device (e.g. bear hugger [3M, USA]) must be used. In addition, the use of a tumescent infiltration containing triamcinolone, ropivacaine, adrenaline, and hyaluronidase on the subgaleal plane,55 both at the incision site and on the regions where dissection is going to be performed, can be of invaluable help in providing hemostasis, analgesia and reducing swelling in these patients. In places where, devices such as cell saving, or medications like erythropoietin or tranexamic acid are not available, the treating surgeons must be extremely alert of silent bleeders.
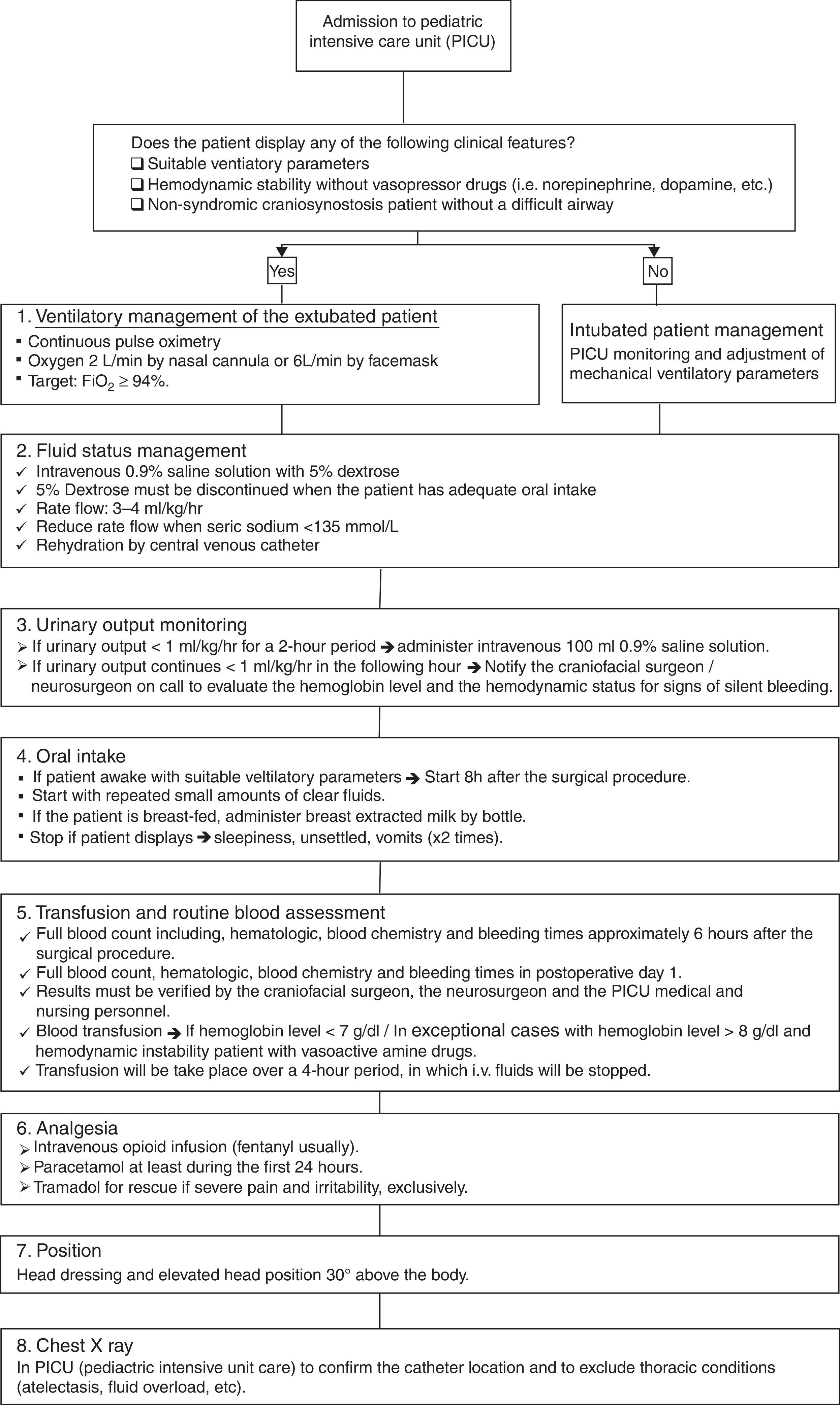
The postoperative treatment protocol (Fig. 2) initiates as the patient is transferred to the pediatric intensive care unit (PICU), where the pediatric anesthetist and the craniofacial surgeon must give a detailed explanation of the findings and technical aspects performed to the PICU medical and nursing personnel. Patients with non-syndromic craniosynostosis and a normal airway, those who demonstrate suitable ventilatory findings and those who are hemodynamically stable without vasoactive drugs can be managed extubated without mechanical ventilatory support. If any of the previous characteristics are present, it is safer to keep the patient intubated, until stability is obtained. The immediate postsurgical management protocol is based on 6 aspects: ventilatory management, fluid support, urinary output monitoring, oral intake, transfusion of blood products, analgesia, position and general measures; and adjuncts (e.g. chest X-ray). The first aspect is the ventilatory management, which should be based on the clinical information displayed by the patient and by continuous pulse oximetry. In extubated patients, oxygen must be administered via a nasal cannula or face mask, aiming a FiO2 higher than 94%. Fluids and hemodynamic status monitoring represent the second aspect, where 0.9% normal saline with 5% dextrose must be used. Once the patient has adequate oral intake the 5% dextrose can be discontinued and the normal saline rates’ reduced. The intravenous fluids will be administered via the central line, with a rate of 3–4ml/kg/h; keeping in mind the blood sodium levels. The third aspect is the urinary output monitoring. If less than 1ml/kg/h for 2 consecutive hours, a bolus of 100ml 0.9% saline must be given. When the patient continues displaying decreased urinary output levels, the craniofacial/neurosurgeon must be notified. The fourth aspect is oral intake. When patients are awake, settled and stable for the 6–8h following the operation, the oral intake with clear fluids in small amounts can be initiated. Patients who are breast-fed, breast milk can be extracted and given. Operated babies and children who display multiple episodes of emesis, are unsettled or show respiratory instability; oral intake can be stopped. Blood product transfusion is based on clinical data and trustable blood tests (complete blood count). Additional packed red blood cells will be given in patients who display anemic signs and present hemoglobin levels <7g/dl. Exceptional cases with hemoglobin level >8g/dl will require blood products. While these products are administered (approximately over 4h), other intravenous fluids should be stopped. Blood products should be transfused via a separate lumen of the central line. The nursing and medical personnel should be very alert for potential allergic reactions to blood products. Pain control should be based on opioid infusion, with paracetamol; using other medications only in the rare event of the patient showing severe pain. The chest X-ray should be performed and verified to assess the pulmonary status and the central venous line location.

Once the patient is transferred to the ward, nursing and medical personnel should keep a constant monitoring of the previous aspects. Keeping in mind that the nurse to patient ratio in public hospitals in developing countries is 1:6–8 and that family members and guardians may have a humble cultural and educational background, it is imperative that constant supervision takes place by the treating surgeon and residents. The patient can be mobilized and kept in different positions to avoid pressure ulcers.
When the patient is settled, eating properly and showing stability, discharge from the hospital can be thought. Parents and guardians must be counseled about being consistent about feeding the baby, keeping in mind the nutritional requirements. The patient can be bathed, including the head, on a daily basis. The use of antibiotic ointment on the skin incisions can prove helpful in certain cases. Paracetamol will be enough to control pain. As with other pediatric patients, spending time to explain the parent/guardian about the features that could raise the suspicion of pain, will be a great investment on the overall patient care. Parents/guardian should be alert for the following alarm signs: temperature of >38°C that cannot be controlled with medication, recurrent episodes of projectile vomiting, the patient being continuously unsettled or with drastic changes in conduct, or the patient waking up unexpectedly at night with constant crying. When any of the previous features are present, the patient should be examined at the emergency room of the treating craniofacial center or seen by the treating doctors as soon as possible.
On postoperative day 10, the patient should be reexamined, looking for persistent fluid collections. Any signs of localized infection, in addition to the oral intake, behavior and level of activity should be assessed on the same visit and at the 4th, 8th, 12th weeks as well as at 6 and 12 months following surgery. Once the patient is 5 years old, the monitoring can continue every year. Keeping in mind that the parents/guardians need a little bit more monitoring than in clinical settings, the treating surgeons must keep a proactive and enquiring conduct.
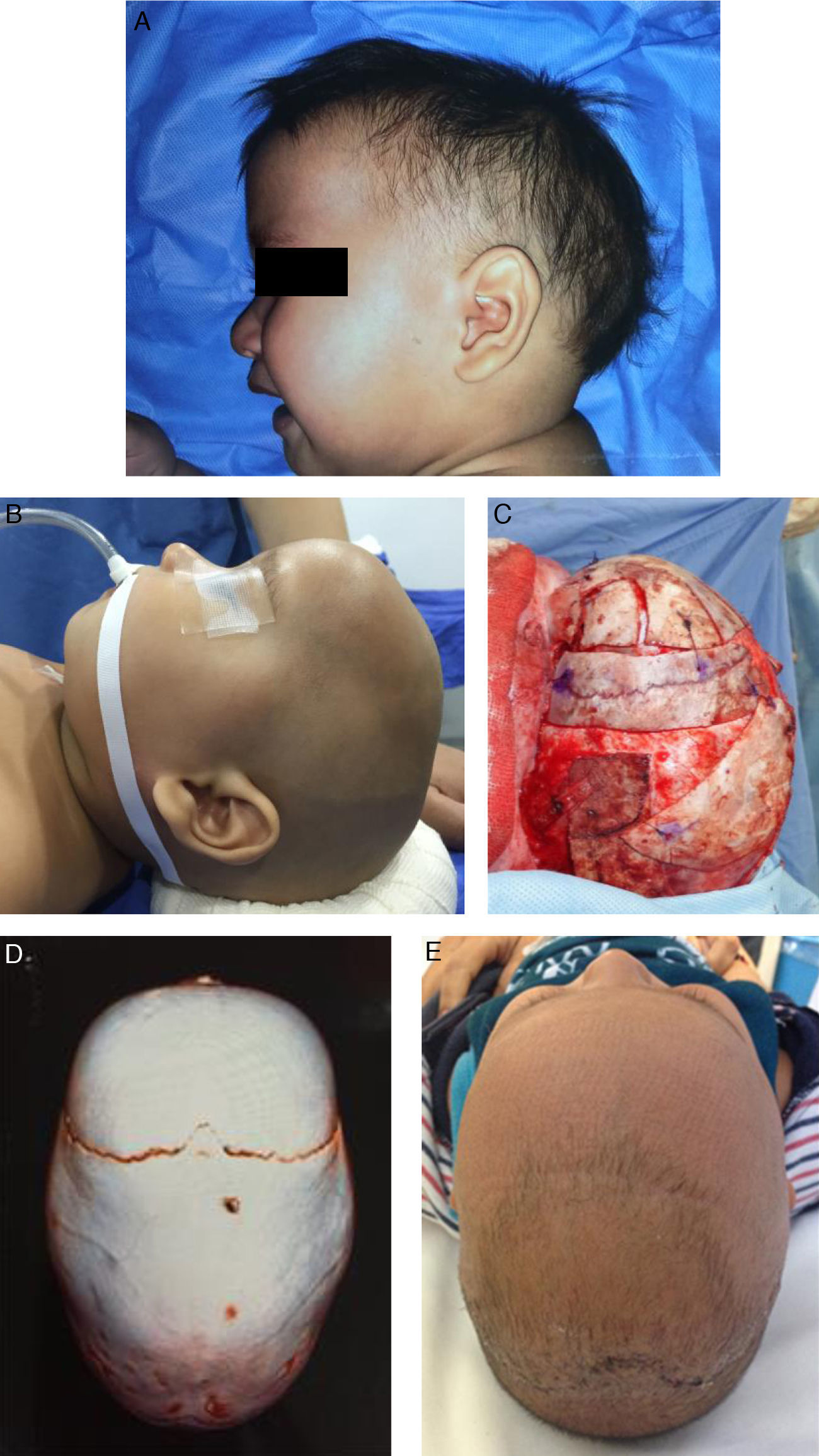
Fig. 3 shows the photos and tomographic images of a 10-month old boy with sagittal craniosynostosis who underwent a total cranioplasty, based on the Melbourne method,39 performed at our institution.
 preoperative lateral view, (B) preoperative lateral view on table, (C) transoperative lateral view on table, after remodeling, (D) preoperative 3D CT, view from above, (E) postoperative view from above at 2-months follow-up.")
A 10-month old boy with sagittal craniosynostosis who underwent a total cranial vault remodeling based on the Melbourne method.39 (A) preoperative lateral view, (B) preoperative lateral view on table, (C) transoperative lateral view on table, after remodeling, (D) preoperative 3D CT, view from above, (E) postoperative view from above at 2-months follow-up.
It is essential to keep adequate communication with the pediatrician following the patient at their hometown, looking for overall body and head growth, cognitive status and psychomotor development, school performance; and conduct and behavior. The ophthalmologist should follow the patient one year after the surgery, and then, if needed, based on suspicious signs of intracranial hypertension. The pediatric psychologist should also be involved to document and compare the development as the patient grows.
ConclusionsThe adequate management of patients with craniosynostosis requires a multidisciplinary team, with a well-organized approach, based on three pillars: the patient's clinical and tomographic features; the child's social and demographic aspects; and the technical expertise and available resources at the treating craniofacial center. The already complex treatment protocol can be even more difficult in developing countries due to social and demographic factors.
It is imperative for the multidisciplinary team to speak the same language by following structured guidelines, in which every single step is carefully explained. We propose to standardize and unify criteria to improve outcomes, optimize resources and get better functional and esthetic results in our patients. With a thorough coordination by the craniofacial clinic, by following the same rules, and via an appropriate staff training, including medical, nursing and technical personnel; potential complications can be avoided.
An excellent and continuous relationship between the treating medical personnel and the parents/guardians is mandatory. Medical and non-medical aspects should be communicated using lay language with the aim of promoting a liaison that will translate in a good postoperative care of the patient.
Ethical disclosureProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestThe authors declare that they have no conflict of interests.
Nunthasiri Wittayanakorn, MD. Pediatric neurosurgery fellow, Neurosurgery department, Royal Children's Hospital Melbourne, 50 Flemington Road, Parkville, Victoria 3052, Australia.
César Athié Gutiérrez MD. CEO, Hospital General de México “Dr. Eduardo Liceaga”, Dr Balmis 148, Col. Doctores, Cuauhtémoc, 06720 Mexico City, Mexico.
Salvador Cuéllar y Martínez, MD. Consultant, Pediatric neurosurgery department, Hospital General de México “Dr. Eduardo Liceaga”, Dr Balmis 148, Col. Doctores, Cuauhtémoc, 06720 Mexico City, Mexico.
Silvia del Carmen Espinosa Maceda, MD. Head of Unit, Plastic and reconstructive surgery department, Hospital General de México “Dr. Eduardo Liceaga”, Dr Balmis 148, Col. Doctores, Cuauhtémoc, 06720 Mexico City, Mexico.
Julio Palacios-Juarez, MD. Plastic and Reconstructive Surgery Resident, Plastic and reconstructive surgery department, Hospital General de México “Dr. Eduardo Liceaga”, Dr Balmis 148, Col. Doctores, Cuauhtémoc, 06720 Mexico City, Mexico.
José de Jesús Gutiérrez Cabrera, MD. Head of unit, Pediatric neurosurgery department, Hospital General de México “Dr. Eduardo Liceaga”, Dr Balmis 148, Col. Doctores, Cuauhtémoc, 06720 Mexico City, Mexico.