





The pattern of structural alterations of the glomerular basement membrane comprises two hereditary glomerular diseases: Alport syndrome (AS) and thin basement membrane nephropathy (TBMN); both are rare entities.
Material and methodsA retrospective, descriptive and observational study was carried out. A sample of 90 cases from Hospital General de México “Eduardo Liceaga” and the Instituto Nacional de Cardiología “Ignacio Chávez” were obtained over a period of 5 years (2011–2016). The diagnoses provided in all cases were reviewed and the clinical and histological spectrum was described. Data were analysed using descriptive statistics.
ResultsStructural alterations of the basement membrane were found to be more frequent in women (60%), and occurred most frequently in the second decade of life. We found 38 cases (42.22%) compatible with TBMN and 52 cases (57.77%) with ultrastructural changes suggestive of Alport syndrome. Of the cases with characteristics compatible with AS, the majority were men (55.76%) with an average age of 14 years (3–39 years), starting with haematuria (42.30%); in the electron microscopy (EM), the glomerular basement membranes (GBM) measured on average 245.27nm, with very irregular ranges. The majority of patients with TBMN were women (81.57%) with an average age of 29 years (6–66 years) and with persistent microscopic haematuria at the time of diagnosis (47.36%); in the EM, the GBM averaged 170.63nm in thickness.
ConclusionsWe report one of the largest case series in Latin America with respect to entities that share a morphological pattern via the study of optical microscopy, that of structural alterations of the glomerular basement membrane. With the EM study, both entities can be suggested.
El patrón de alteraciones estructurales de la membrana basal glomerular comprende a dos enfermedades hereditarias del glomérulo: el síndrome de Alport (SA) y la enfermedad de membranas basales delgadas (EMBD); ambas son entidades poco frecuentes.
Material y métodosSe hizo un estudio retrospectivo, descriptivo y observacional. Se obtuvo una muestra de 90 casos del Hospital General de México “Eduardo Liceaga” y del Instituto Nacional de Cardiología “Ignacio Chávez” de un período de 5 años (2011 a 2016). Se revisaron los diagnósticos prestados en todos los casos y se describió el espectro clínico e histológico. Los datos fueron analizados utilizando estadística descriptiva.
ResultadosSe encontró que las alteraciones estructurales de la membrana basal eran más frecuentes en las mujeres (60%), y la mayor parte se presentaron en la segunda década de la vida. Se encontraron 38 casos (42.22%) compatibles con enfermedad de membranas basales delgadas (EMBD) y 52 casos (57.77%) con cambios ultraestructurales sugerentes de síndrome de Alport. De los casos con características compatibles con SA, la mayoría eran hombres (55.76%) con una edad promedio de 14 años (3–39 años), debutando con hematuria (42.30%); en el estudio de microscopia electrónica de transmisión (MET) las membranas basales glomerulares (MBG) midieron en promedio 245.27nm, con rangos muy irregulares. La mayoría de los pacientes con EMBD eran mujeres (81.57%) en una edad promedio de 29 años (6–66 años), con hematuria microscópica persistente al momento de su diagnóstico (47.36%); en el estudio de MET las MBG en promedio midieron 170.63nm de grosor.
ConclusionesSe informa una de las casuísticas más grandes de Latinoamérica respecto a entidades que comparten un patrón morfológico por el estudio de microscopía óptica, el de alteraciones estructurales de la membrana basal glomerular. Con el estudio de MET se pueden sugerir ambas entidades.
Alport syndrome (AS) and thin basement membrane disease (TBMD) are genetically heterogeneous diseases characterised by structural abnormalities in the glomerular basement membrane (GBM).1 They are entities that do not show specific findings in the optical microscopy study for their diagnosis, so it can be difficult to differentiate between them. The current recommendations suggest the genetic study as the gold standard for the diagnosis of AS and to exclude it in a case of TBMD2; however, by using extension studies such as direct immunofluorescence and transmission electron microscopy (TEM), it is possible to diagnose, or at least to suggest, these disorders in most cases.3
AS is currently considered a spectrum of pathologies secondary to mutations in the α3, α4 and α5 chains of type IV collagen, usually with auditory and ocular abnormalities. Its incidence in the US is 1:5000–1:10,000, according to the database of the different mutations.4 Approximately 85% of cases are due to several mutations in the COL4A5 gene, located on chromosome Xq26-48, which encodes the α5 chain of type IV collagen, presenting progressive kidney disease until terminal stages in affected men but only with urinary disorders (haematuria with or without mild proteinuria, which may increase with age) in women.5 Although most patients with X-linked AS will have a family history of kidney disease, 10%–15% of cases represent de novo mutations in the COL4A5 gene. The remaining AS cases result from mutations in the COL4A3/COL4A4 locus on chromosome 2q35-37, which encode the α3 and α4 chains of type IV collagen with autosomal recessive inheritance; autosomal dominant inheritance is less common. The phenotypic expression of these patients is quite variable, ranging from benign urinary disorders to progressive nephropathy. Ultrastructural abnormalities in the MBG may be less severe in autosomal recessive than in X-linked, with a predominance of thinned GBM (similar to the changes observed in TBMD or heterozygous carriers of X-linked AS) rather than extensive lamination. Therefore, it is possible that some patients reported with TBMD who developed hypertension, proteinuria and late-onset renal failure may have autosomal recessive AS with homozygous or heterozygous mutations of COL4A3/COL4A4.6 Mutations in the alpha 3, alpha 4 or alpha 5 chains of type IV collagen in AS result in a defective assembly of the heterotrimer, which is essential for the GBM function, the lens of the eye and the cochlea in the ear. This results in the classic clinical manifestations of established AS.7 Patients present with haematuria from infancy. Proteinuria develops during adolescence with nephrotic syndrome in 30%–40%. Progressive deafness develops in approximately half of adult men with Alport syndrome, with ocular defects in about one-third, most commonly anterior lenticonus.8
TBMD is the underlying morphological disorder in most families diagnosed with benign familial haematuria and can also be observed in sporadic cases of haematuria. Patients show persistent microscopic haematuria, often with intermittent macroscopic haematuria. Of the patients biopsied due to persistent isolated haematuria, TBMD may be present in 20%–25%. The prognosis is generally benign, with a slight increase in the risk of chronic kidney disease in AS carriers.9 Approximately 40% of patients with TBMD have heterozygous mutations at the COL4A3/COL4A4 locus, and can therefore be considered carriers for autosomal recessive AS.10 In addition, two mutations (G1334E and G871C) were recently identified as being associated with a form of TBMD characterised by the development of focal and segmental glomerulosclerosis and proteinuria after 30 years and kidney failure after 50 years of age.11 This further blurred the dividing line between TBMD and AS. TBMD appears to be much more common than AS, occurring in approximately 1% of the population compared with 0.02% for AS.12 However, as noted above, there is considerable phenotypic overlap between individuals, especially children and young adults.13
Despite the low frequency of GBM structural disorders, many of them are never diagnosed due to the late care of patients arriving with advanced stage nephropathies. It is necessary to report the cases reviewed with these pathologies to learn about a representative sample of the Mexican population and to characterise each of them, since it is the first step so that in the future decisions will be taken both in the follow-up and in their treatment. The objectives of this study are to report the cases reported with a pattern of GBM structural disorders, to classify or suggest the type of structural alteration of the glomerular basement membrane (AS, TBMD) according to the TEM study and to characterise the clinical and histopathological data of the cases studied.
Material and methodsA retrospective, descriptive and observational study was conducted. The records of a 5-year period from 1 January 2011 to 1 December 2016 that were reported as GBM structural disorders or compatible with this diagnosis were obtained by consecutive non-probabilistic sampling using the database of the biopsies registry of the Surgical Pathology Department of Hospital General de México “Eduardo Liceaga” and the “Ignacio Chávez” Instituto Nacional de Cardiología. Of a total of 105 cases, 8 cases were excluded that did not have a TEM study, which was not performed because it was a suboptimal sample or due to the inadequacy of the material for the processing of this study. Seven cases were eliminated in which the structural disorders were not specific to GBM, most of them presenting advanced healing data that were attributed to adaptive processes of another pathology or constitutional type. To classify each case, a minimum of 10 glomerular capillary loops were examined ultrastructurally. The thickness of the GBM was measured on all capillary loops and at least 30 readings, which evenly covered the circumference of the loops, were recorded. The average and thickness range of the GBMs were also documented. A normal thickness of 320–340nm was established for GBM in adults and for those under 11 according to values adjusted for age and sex according to the reported standards.1 The clinical and histopathological spectrum was described in each one. The data were analysed using descriptive statistics. The results are presented as numbers, percentages and averages.
ResultsOf the 90 cases studied, a total of 36 men (40%) and 54 women (60%) were used. The age range found was very broad, from 3 years to 66 years. The age distribution was divided into age groups, each corresponding to one decade of life. The largest number of cases (35.55%) occurred in the second decade of life (11–20 years) followed by the first decade (28.88%). In order to separate the two large entities that share the morphological pattern of GBM structural disorders, TBMD and ultrastructural changes compatible with AS, the TEM findings were used, since in our environment there is no specific immunoreactant for each one of the type IV collagen chains to classify each of the variants. In AS, the TEM findings are the multilamination of the GBM dense lamina, which confers an “interwoven” or “basket-like” appearance. In addition, it is possible to observe microparticles in the GBM or “bread crumbs” between the laminations, a scalloped section or “excrescence” of the subepithelial surface of the GBM and a variable and irregular thickness, both thick and thin GBMs, unlike TBMD, where thinning is uniform and homogeneous (>50% of GBMs) and measure <200nm in thickness. In this way, 38 cases (42.22%) were compatible with TBMD and 52 cases (57.77%) with ultrastructural changes suggestive of AS.
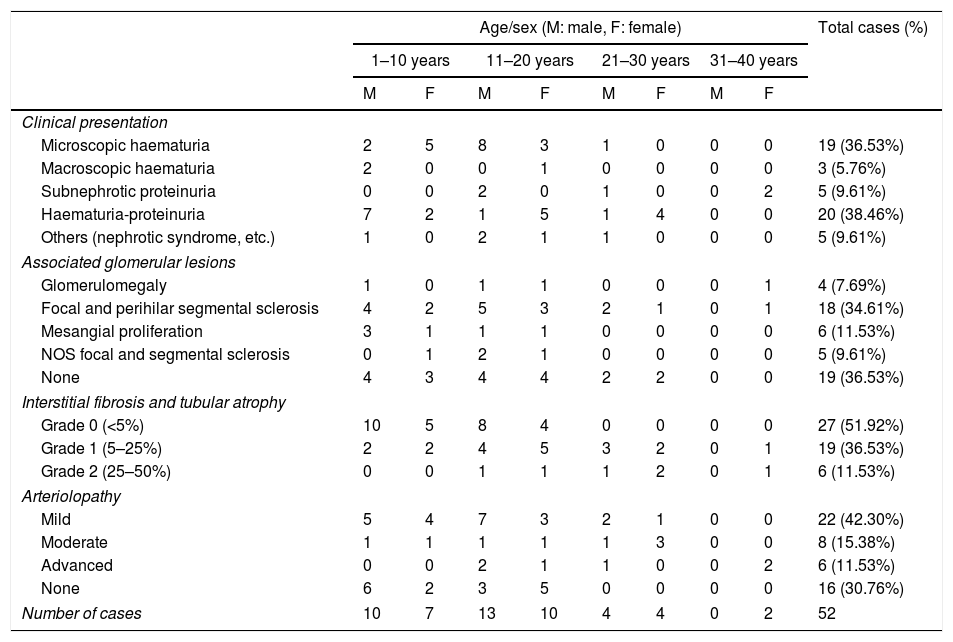
Of the 52 cases with characteristics compatible with AS, the majority were men (29 cases, 55.76%). The age of the patients ranged from 3 to 39 years; the majority (23 cases) were detected in the second decade of life (44.23%), with a mean of 14 years. The findings on clinical presentation, glomerular lesions overloaded with the pattern of structural changes in GBM, degree of interstitial fibrosis and associated tubular atrophy and degree of arteriolopathy are shown in Table 1. It should be noted that a history of auditory and/or visual alterations (neurosensorial deafness, corneal opacities, etc.) were reported in only 5 cases (9.61%). In the histopathological study of the cases with characteristics compatible with AS, minimal changes were observed in the glomeruli. Most of them were anodyne (36.53%), with folded and laminated GBM segments, alternating with others with “thin” appearance and only 6 cases (11.53%) showed mild mesangial hypercellularity, especially in younger patients. The interstitial foamy macrophages were only present in those cases with a history of proteinuria. The direct immunofluorescence study was negative for all the immunoreactants that are routinely analysed (IgG, IgA, IgM, C1q, C3c, C4c, Fibrinogen, Albumin, Kappa and Lambda). Deposits were only detected by entrapment in those glomeruli with zones of segmental sclerosis (non-specific deposit). On average, the GBMs of cases with AS measured 245.27nm, a lower than expected average for the normal thickness of glomerular basement membranes in adults. However, it is necessary to take into account the number of patients under 11 years of age and the heterogeneous nature of the measurements, since an average minimum measurement of 132.16nm and an average maximum measurement of 464.96nm were found, with measurements as thin as 57nm to thick segments of 1039nm. This explains the irregularity of the membranes in this entity (Fig. 1) and by definition they cannot be pigeonholed as TBMD.
Characteristics of the cases with ultrastructural changes suggestive of Alport syndrome.
| Age/sex (M: male, F: female) | Total cases (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1–10 years | 11–20 years | 21–30 years | 31–40 years | ||||||
| M | F | M | F | M | F | M | F | ||
| Clinical presentation | |||||||||
| Microscopic haematuria | 2 | 5 | 8 | 3 | 1 | 0 | 0 | 0 | 19 (36.53%) |
| Macroscopic haematuria | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 3 (5.76%) |
| Subnephrotic proteinuria | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 2 | 5 (9.61%) |
| Haematuria-proteinuria | 7 | 2 | 1 | 5 | 1 | 4 | 0 | 0 | 20 (38.46%) |
| Others (nephrotic syndrome, etc.) | 1 | 0 | 2 | 1 | 1 | 0 | 0 | 0 | 5 (9.61%) |
| Associated glomerular lesions | |||||||||
| Glomerulomegaly | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 4 (7.69%) |
| Focal and perihilar segmental sclerosis | 4 | 2 | 5 | 3 | 2 | 1 | 0 | 1 | 18 (34.61%) |
| Mesangial proliferation | 3 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 6 (11.53%) |
| NOS focal and segmental sclerosis | 0 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 5 (9.61%) |
| None | 4 | 3 | 4 | 4 | 2 | 2 | 0 | 0 | 19 (36.53%) |
| Interstitial fibrosis and tubular atrophy | |||||||||
| Grade 0 (<5%) | 10 | 5 | 8 | 4 | 0 | 0 | 0 | 0 | 27 (51.92%) |
| Grade 1 (5–25%) | 2 | 2 | 4 | 5 | 3 | 2 | 0 | 1 | 19 (36.53%) |
| Grade 2 (25–50%) | 0 | 0 | 1 | 1 | 1 | 2 | 0 | 1 | 6 (11.53%) |
| Arteriolopathy | |||||||||
| Mild | 5 | 4 | 7 | 3 | 2 | 1 | 0 | 0 | 22 (42.30%) |
| Moderate | 1 | 1 | 1 | 1 | 1 | 3 | 0 | 0 | 8 (15.38%) |
| Advanced | 0 | 0 | 2 | 1 | 1 | 0 | 0 | 2 | 6 (11.53%) |
| None | 6 | 2 | 3 | 5 | 0 | 0 | 0 | 0 | 16 (30.76%) |
| Number of cases | 10 | 7 | 13 | 10 | 4 | 4 | 0 | 2 | 52 |

Segments of glomerular basement membrane from a case with Alport syndrome seen by electron microscopy at higher magnification. It shows splitting and multilamination with a classic “weave” or “basket-like” appearance; it also contains microparticles “in bread crumbs”. TEM, 20,000×.
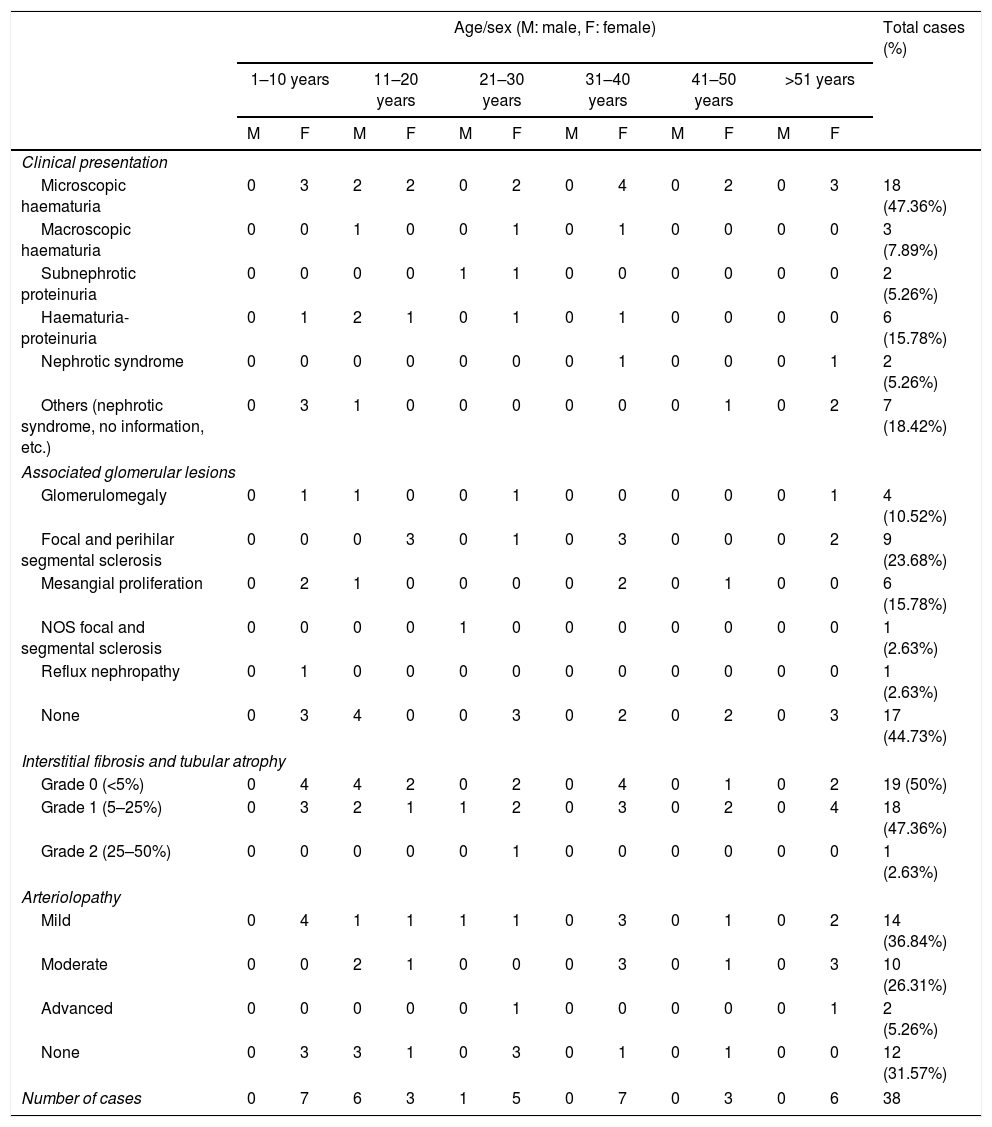
Of the 38 patients with TBMD, the vast majority were women (81.57%) and only 7 (18.42%) were men. The age range was from 6 to 66 years, with a mean of 29 years, being more common in the second decade of life (23.68%). The findings on clinical presentation, glomerular lesions overloaded with the pattern of GBM structural changes, degree of interstitial fibrosis and associated tubular atrophy and degree of arteriolopathy are shown in Table 2. In the histopathological study, it was observed that in the majority of cases (42.10%) the glomeruli were within the normal limits. Some segments of glomerular basement membranes with “thinning” appearance were observed alternating with dilated capillary loops (Fig. 2), some with erythrocytes in their lumens and pigmented cylinders in the tubules. There was focal widening of the mesangial matrix with mild proliferation in only 6 cases (15.78%). As in AS, the direct immunofluorescence study did not have specific deposits in any of the cases. However, routine immunoreactants are performed to rule out other pathologies that have the same clinical presentation, such as IgA nephropathy. Finally, the most important findings to define the TBMD were found in TEM, on average the GBMs measured 170.63nm (Fig. 3), with an average minimum measurement of 92.52nm and an average maximum measurement of 272.84nm.
Characteristics of cases compatible with thin basement membrane disease.
| Age/sex (M: male, F: female) | Total cases (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1–10 years | 11–20 years | 21–30 years | 31–40 years | 41–50 years | >51 years | ||||||||
| M | F | M | F | M | F | M | F | M | F | M | F | ||
| Clinical presentation | |||||||||||||
| Microscopic haematuria | 0 | 3 | 2 | 2 | 0 | 2 | 0 | 4 | 0 | 2 | 0 | 3 | 18 (47.36%) |
| Macroscopic haematuria | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 3 (7.89%) |
| Subnephrotic proteinuria | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (5.26%) |
| Haematuria-proteinuria | 0 | 1 | 2 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 6 (15.78%) |
| Nephrotic syndrome | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 2 (5.26%) |
| Others (nephrotic syndrome, no information, etc.) | 0 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 2 | 7 (18.42%) |
| Associated glomerular lesions | |||||||||||||
| Glomerulomegaly | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 4 (10.52%) |
| Focal and perihilar segmental sclerosis | 0 | 0 | 0 | 3 | 0 | 1 | 0 | 3 | 0 | 0 | 0 | 2 | 9 (23.68%) |
| Mesangial proliferation | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 6 (15.78%) |
| NOS focal and segmental sclerosis | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.63%) |
| Reflux nephropathy | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.63%) |
| None | 0 | 3 | 4 | 0 | 0 | 3 | 0 | 2 | 0 | 2 | 0 | 3 | 17 (44.73%) |
| Interstitial fibrosis and tubular atrophy | |||||||||||||
| Grade 0 (<5%) | 0 | 4 | 4 | 2 | 0 | 2 | 0 | 4 | 0 | 1 | 0 | 2 | 19 (50%) |
| Grade 1 (5–25%) | 0 | 3 | 2 | 1 | 1 | 2 | 0 | 3 | 0 | 2 | 0 | 4 | 18 (47.36%) |
| Grade 2 (25–50%) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.63%) |
| Arteriolopathy | |||||||||||||
| Mild | 0 | 4 | 1 | 1 | 1 | 1 | 0 | 3 | 0 | 1 | 0 | 2 | 14 (36.84%) |
| Moderate | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 3 | 0 | 1 | 0 | 3 | 10 (26.31%) |
| Advanced | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 2 (5.26%) |
| None | 0 | 3 | 3 | 1 | 0 | 3 | 0 | 1 | 0 | 1 | 0 | 0 | 12 (31.57%) |
| Number of cases | 0 | 7 | 6 | 3 | 1 | 5 | 0 | 7 | 0 | 3 | 0 | 6 | 38 |
 showing a normal glomerulus, typical of TBMD in optical microscopy. Only some dilated capillary loops are striking. PAS staining, 40×.")

The pattern of GBM structural disorders classically defines two entities that share characteristics in the optical microscopy study, AS and TBMD. In both, almost normal glomeruli can be observed; some differences found in this study were that in cases compatible with AS, the MBG GBMs predominate with others of “thin” appearance and in the TBMD, in addition to segments of thin GBMs, they tend to form ectasia of capillaries with erythrocytes in their lumens. Despite this, the findings are very subtle and may go unnoticed by an inexperienced pathologist, and this, coupled with overlapping clinical data (such as age and presentation with haematuria) makes it difficult to catalogue each one. As reported in some cases,14 the results of the biopsy may confirm the diagnosis, but it may also not be conclusive. However, using extension studies allows us to suggest these entities, and with this result it is the duty of the attending physician to confirm the diagnosis through the family history and with the genetic study, which is currently the gold standard for diagnosis.2
In agreement with the international literature, the frequency of these pathologies is low, and microscopic haematuria is the most common presentation. The AS-compatible cases had a broader clinical spectrum, including subnephrotic proteinuria. Consequently, as stated by other authors,15 in the histopathological study more late healing lesions such as focal and segmental sclerosis, interstitial fibrosis and arteriolosclerosis were observed. However, the presence of interstitial foamy macrophages was unusual; only in cases with a history of proteinuria, contradicting what was previously stated about its frequent finding in these specimens.1,8 A younger age of onset was observed, different from that which was referred to in the classic books,16 and it is similar to a recently published series of cases in Asia.17 The morphometry of GBM in Alport syndrome cases had a lower-than-expected average thickness for adults. However, it should be noted that most of the studies conducted to determine the normal thickness of GBM18 are not conducted in the Latin American population, particularly in the Mexican population, which has an average height that is smaller than the Anglo-Saxon population. This together with particular situations, such as low birth weight or different mutations19 determine a GBM thickness that is lower than that of other races. For cases of TBMD, a similarity was found in the clinicopathological characteristics, as reported in the literature.20 However, an association with focal sclerosing and segmental perihilar lesions was observed in some cases, which was considered infrequent,16 but which may be related to new mutations described for TBMD.11
In conclusion, one of the largest samples in Latin America on GBM structural alterations was reported. Although the dividing line between AS and TBMD is increasingly blurred with phenotypic and genetic overlap between patients, which is therefore why they are considered within the same pathological spectrum under the name of “type IV collagen nephropathy”,13 the role of the pathologist remains very important, as not all patients have access to the genetic study. Also, as the current guidelines state,2 it is necessary to continue to separate them by their different management; of course, having as many diagnostic tools as possible, such as TEM, will ensure a greater likelihood of diagnostic accuracy.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article
FundingExisting funds in the Hospital.
Conflict of interestIt is stated that there is no conflict of interest in the publication and/or authorship of this article.
The authors are grateful for the supply of cases from the “Ignacio Chávez” Instituto Nacional de Cardiología.