


La asociación entre síndrome de Down y esclerosis tuberosa (SD/ET) muy pocas veces ha sido descrita. Estas 2 entidades presentan en común una sobreactivación del sistema m-TOR (ligando para rapamicina mamífero-específico), la que se ha visto relacionada con crecimiento neuronal anómalo, transducción sináptica anómala y sobreestimulación de neurotransmisores en diferentes áreas cerebrales, entre otras, que se manifiestan en retardo cognitivo y síntomas autistas en los pacientes en quienes se presentan.
Resumen del casoPresentamos el caso de una niña con diagnóstico de SD, quien comenzó a temprana edad con crisis convulsivas tónico-clónicas y placas hipopigmentadas en piel. Se practicó una resonancia cerebral, la cual evidenció imágenes de túberes corticales sugestivas de ET. Un ecocardiograma posterior evidenció la presencia de múltiples lesiones endomiocárdicas sugestivas de rabdomiomas. Controles ulteriores mostraron resolución espontánea de estas lesiones cardiacas, pero con persistencia de las alteraciones cerebrales. Se describe la refractariedad de los episodios convulsivos a pesar de tratamiento apropiado con múltiples anticonvulsivantes.
ConclusionesLas implicaciones neurológicas y cardiacas en pacientes con asociación SD/ET ocupan un escenario terapéutico difícil, con alta tasa de complicaciones, en parte debidas a la refractariedad sintomática. Los nuevos medicamentos dirigidos hacia la vía de señalización m-TOR podrían ser de gran utilidad en este escenario, dada su relación con la mejoría del deterioro cognitivo y las tumoraciones asociadas a estas 2 entidades.
The association between Down syndrome and Tuberous sclerosis (DS/TS) had been very rarely described. These two entities present in common overactivation of m-TOR system (mammalian target of rapamycin), which has been related to abnormal neuronal growth, abnormal synaptic transduction and overstimulation of neurotransmitters in different brain areas, among others, that are manifested in cognitive retardation and autistic symptoms.
Case summaryWe present a girl with diagnosis of DS who debuted one early childhood with convulsive tonic-clonic seizures and hypopigmented skin plaques. A brain MRI which reported cortical tubers suggestive of TS. A subsequent echocardiogram showed the presence of multiple lesions suggestive of endomyocardial rhabdomyomas. Further medical checks showed spontaneous resolution of these cardiac lesions, but persistence of brain disorders. We also describe the refractoriness of the convulsive events despite appropriate treatment with multiple anticonvulsants.
ConclusionsNeurological and cardiac implications in patients with DS/TS association dealing a difficult therapeutic stage high rate of complications, in part due to refractory symptomatic. New drugs directed towards the signaling pathway m-TOR could be very useful in this scenario, as it relates to improvement of cognitive impairment and tumors associated with these two entities.
El síndrome de Down (SD), es considerado la principal causa de retardo mental de origen cromosómico1,2, con una prevalencia de 1:150 concepciones y 1:3.000 nacidos vivos, esto debido a que gran parte de estas fecundaciones son inviables. Por otra parte, la esclerosis tuberosa (ET) se caracteriza por la transmisión autosómica dominante de la inactivación de los genes TSC1 o TSC2 (cromosomas 9 y 16)3, los cuales codifican para las proteínas hamartina y tuberina, respectivamente; con una prevalencia de 1:10.000 nacimientos; está enmarcado dentro de los síndromes neurocutáneos4, afectando a la piel y al sistema nervioso central (SNC) con adenomas sebáceos, máculas hipopigmentadas, fibromas periungueales de Koenen, angiomiolipomas, túberes corticales, nódulos subependimarios y rabdomiomas cardiacos, entre otros5.
La aparición concomitante SD/ET ha sido comunicada de manera anecdótica. Estudios previos han sugerido que estos síndromes no tienen relación causal/clínica y que su presentación es el producto de la prevalencia de cada condición por separado (1:30.000.000 nacimientos)6,7, por lo que resulta interesante poder conocer los detalles del comportamiento clínico de esta asociación, y si existen similitudes en vías moleculares específicas que puedan mostrar patrones génicos y moleculares en común, como es el caso de la vía de señalización m-TOR, la cual se encuentra sobreexpresada en estos pacientes y condiciona crecimiento celular anómalo y deterioro cognitivo, situación que se detallará más adelante.
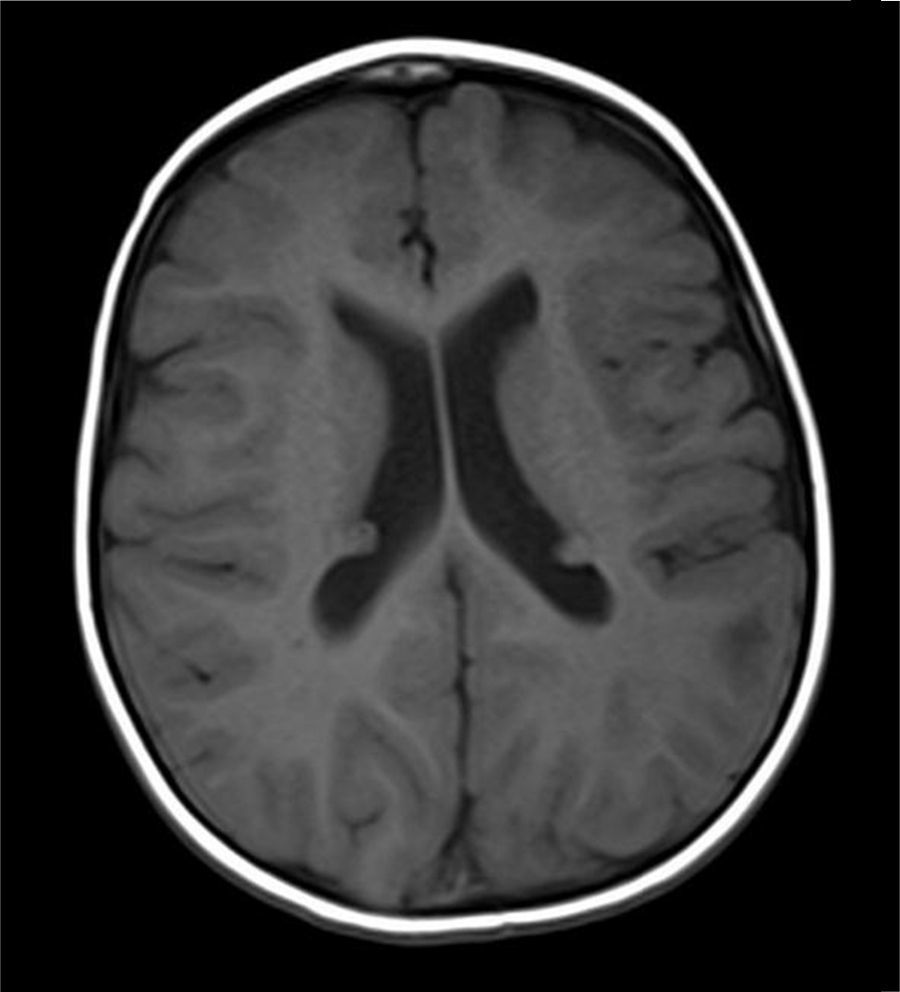
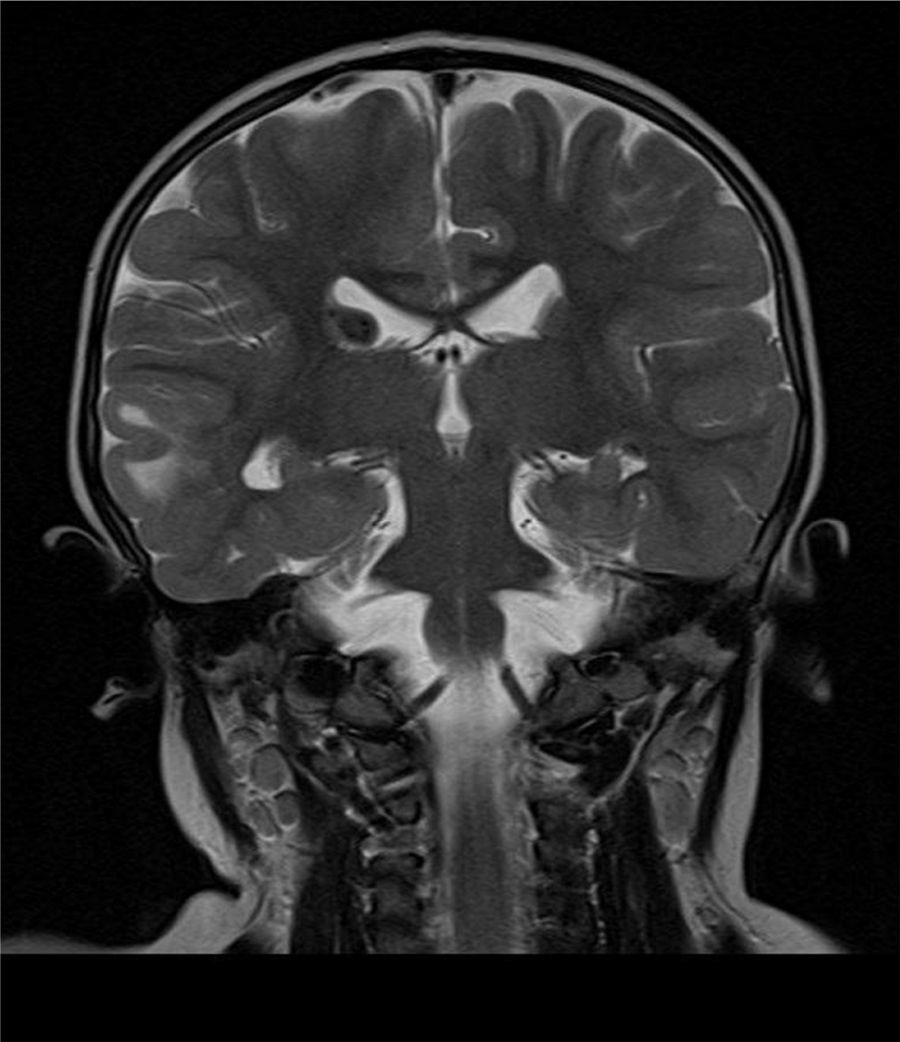
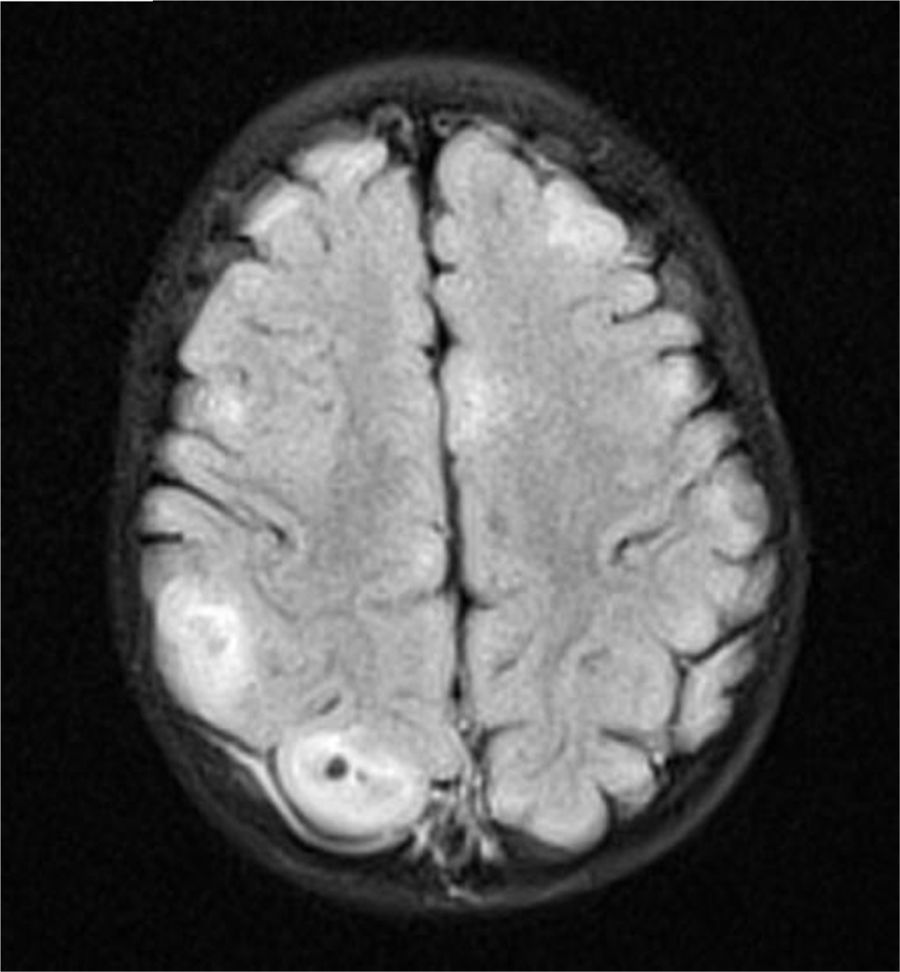
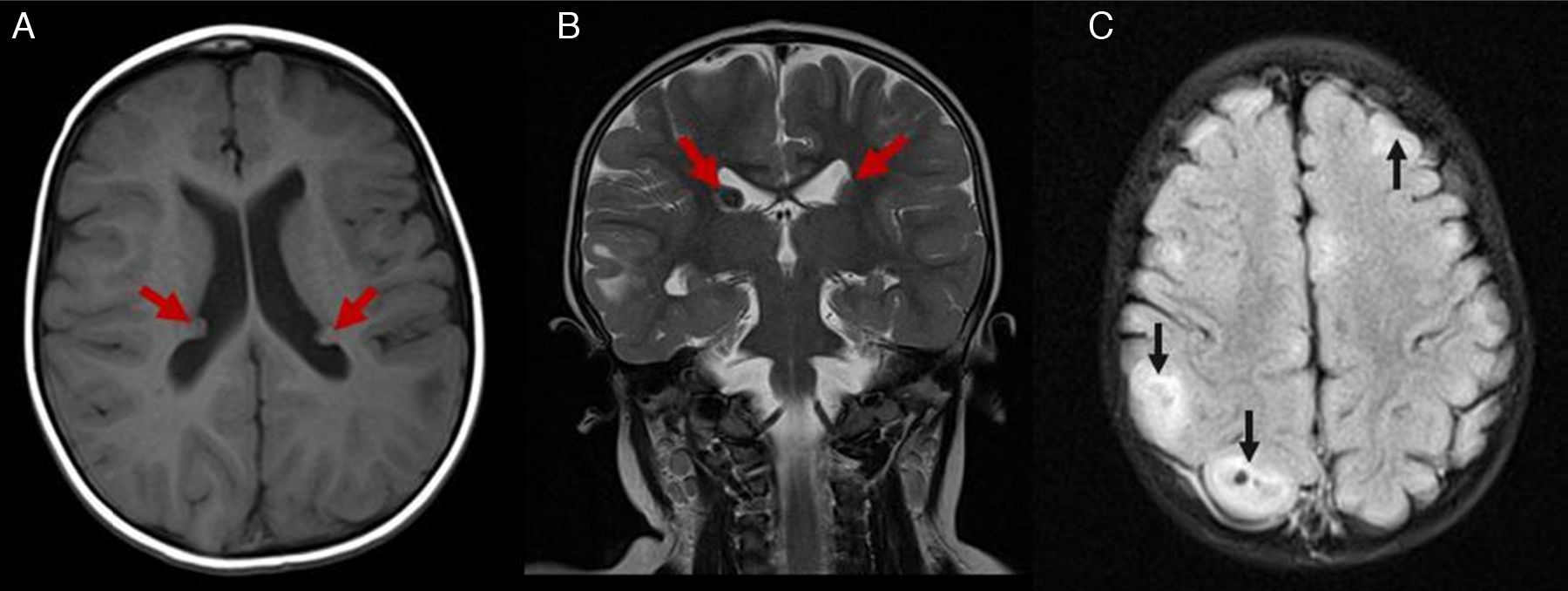
Caso clínicoUna niña nacida a los 8 meses de edad gestacional, con rasgos y cariotipo confirmatorios de SD (47XX+21; cariotipo normal en los padres), sin antecedentes importantes durante su gestación, presentó como hallazgo ecocardiográfico un foramen oval permeable restrictivo con la presencia de al menos 3 masas intramiocárdicas de 5×5mm en promedio, en paredes ventriculares y septo interventricular, compatibles con rabdomiomas, sin compromiso hemodinámico. Debido a la asociación entre rabdomiomas cardiacos y ET, se realizó una tomografía axial computarizada (TAC) cerebral, la cual mostró imágenes densas en la sustancia blanca subcortical y subependimaria correspondientes a túberes corticales. La resonancia magnética nuclear (RMN) cerebral con contraste mostró lesiones corticales múltiples y subependimarias (figs. 1–3) concordantes con nódulos corticales y túberes subependimarios con signos de pérdida de volumen en la fosa posterior (fig. 4).



. C: RMN-FLAIR-Túberes corticales (flechas).")
Semanas después, la paciente comenzó con crisis tónico-clónicas repetitivas y de difícil control con el manejo inicial; el primer electroencefalograma mostró la presencia de actividad epileptiforme fronto-centro-temporal derecha. En una valoración posterior (2 años de edad) se encontraron múltiples manchas hipopigmentadas/hipocrómicas en tronco e hipotiroidismo franco. La paciente permaneció asintomática desde el punto de vista cardiovascular y los controles posteriores ecocardiográficos mostraron la persistencia del foramen oval con resolución espontánea de las lesiones intracardiacas (4 años de edad), al tiempo que se denotaba retardo pondoestatural (talla 92cm; peso 13,6kg) y psicomotor intensos (gateo y sedestación con ayuda; sonidos guturales).
Al momento de esta comunicación (5 años de edad), se continuó la suplencia tiroidea y múltiples anticonvulsivantes, sin verdadero control de sus cuadros convulsivos. Durante los últimos 2 años, había presentado cuadros neumónicos de repetición que la obligaban a recibir tratamiento hospitalario.
DiscusiónNuestro paciente es uno de los pocos casos publicados con asociación SD/ET; con una probabilidad de 1/30 millones de nacidos vivos; los primeros casos fueron comunicados en Italia y Canadá, pero ningún caso había sido descrito en Suramérica hasta este momento. Las opiniones sobre si existe una asociación netamente casual o si hay una conexión genética más profunda, aún están divididas. Se ha descrito una hiperactividad en la vía de señalización m-TOR (ligando de mamíferos específico para rapamicina) en el hipocampo de modelos murinos para SD y ET por aparte, la cual podría dar origen a una anomalía en la migración neuronal y crecimiento atípico de las dendritas, generando anormalidades en las vías de señalización por medio de neurotransmisores como el glutamato o GABA, ocasionando disregulaciones en la plasticidad neuronal y, por consiguiente, manifestándose con alteraciones en los procesos cognitivos, síntoma común y característico de la mayoría de pacientes con SD o ET. Estos procesos cognitivos han tenido mejoría, en modelos animales, gracias a la utilización de terapias farmacológicas dirigidas específicamente a la inactivación de la vía m-TOR por medio de la rapamicina y sus derivados (rapalogos) como el sirolimus o everolimus.
Al menos en el caso de pacientes con ET, se ha demostrado que esta terapia farmacológica produce mejoría en el aprendizaje, y desde hace algunos años se ha venido usando en EE.UU. el everolimus (aprobado por la FDA) para el tratamiento del astrocitoma subependimario de células gigantes (SEGA) con buenos resultados durante su uso continuo8,9.
Dado que los pacientes con ET tienen una aumentada prevalencia de crecimientos celulares anómalos en localizaciones diversas (SNC e intracardiacos), de comportamiento benigno y generalmente sensible a la terapia farmacológica con rapalogos, este podría ser un inicio en el reconocimiento y mejoramiento de las condiciones de vida de los paciente con SD y ET, ya que se podrían mejorar las capacidades intelectuales y los crecimientos tumorales cerebrales (posibles focos epileptogénicos), si se aplican cada vez a más temprana edad; teniendo siempre presente que estas terapias emergentes conllevan numerosos efectos secundarios como infecciones por inmunosupresión, mucositis e insuficiencia renal, entre otras.
ConclusionesActualmente el tratamiento de pacientes con asociación SD/ET se limita a su control sintomático y a una actitud conservadora y expectante. El SD y la ET comparten alteraciones moleculares comunes sobre los procesos dependientes del sistema m-TOR, responsables del daño neuronal y la aparición de tumores benignos. Basados en estos nuevos conocimientos sobre la disregulación en el crecimiento celular en distintos órganos y la sobreestimulación de la actividad neuronal dependiente de la vía m-TOR, es válido pensar que estos pacientes podrían beneficiarse de recibir precozmente estas terapias emergentes con acción inhibitoria sobre esta vía (everolimus o sirolimus).
Se requieren con relativa urgencia estudios que analicen el impacto de estas terapias en estos síndromes por separado, al igual que en el caso de la asociación SD/ET, como una opción más específica encaminada a reducir la morbimortalidad, el deterioro cognitivo y la refractariedad a la terapéutica actual.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.