


La diabetes mellitus es una de las enfermedades más desafiantes del siglo xxi. La Fundación Internacional de Diabetes estimó que más de 371 millones de personas tenían diabetes en el año 2012, y se espera que este número aumente a 552 millones en 20301. A pesar de su alta prevalencia, no más del 25% de los factores de riesgo cardiovascular son conocidos (por ejemplo, el tabaquismo, la hipertensión y la dislipidemia aterogénica), por lo que otros factores como los productos finales de glicación avanzada (AGE) y el estrés oxidativo pueden estar involucrados2. Con la finalidad de aclarar algunos aspectos relacionados con la formación de los AGE y su implicación en las complicaciones cardiovasculares se hace una descripción de la estructura y la formación de estos compuestos en el organismo.
MétodosSe realizó una búsqueda computarizada de artículos en la base de datos de PubMed utilizando los términos MeSH «diabetes mellitus», «productos finales de glicación avanzada», «receptor para productos finales de glicación avanzada» y «enfermedades cardiovasculares». La búsqueda se enfocó en artículos completos en idioma inglés, considerando las publicaciones en el periodo comprendido entre 2005 y 2015.
Formación endógena de productos finales de glicación avanzadaLos AGE son un grupo heterogéneo de compuestos generados a través de la glicación no enzimática de proteínas, lípidos y ácidos nucleicos. Los cambios químicos estructurales que dan lugar a estos compuestos a menudo tardan meses o años, por lo que las proteínas y otras sustancias que tienen una vida media larga son más susceptibles a ser modificadas por la exposición a la glucosa; entre estas se incluyen las proteínas de la matriz extracelular, la mielina, el cartílago y las proteínas del cristalino.
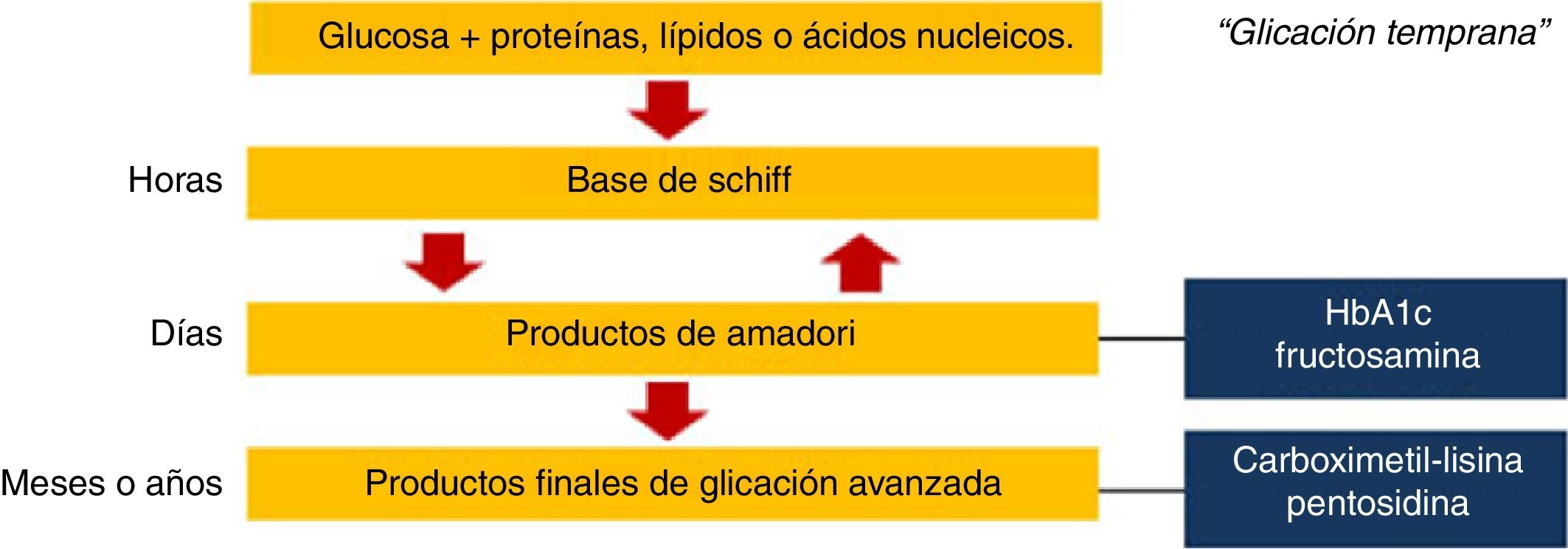
La reacción química inicial denominada «glicación temprana» ocurre entre azúcares reductores o sus productos derivados y los grupos amino en proteínas, lípidos o ácidos nucleicos. Los compuestos originados en esta reacción reversible son inestables y se denominan bases de Schiff3. Posteriormente se someten a un reordenamiento estructural para formar productos de Amadori, siendo los más conocidos la hemoglobina glucosilada y la fructosamina. Sin embargo, ninguno de estos es AGE; para que esto ocurra, los productos de Amadori deben someterse a una mayor deshidratación, oxidación, reacciones de transposición y de fragmentación4. Algunos de los AGE mejor caracterizados son carboximetil, carboximetil-lisina y pentosidina5 (fig. 1).

En el organismo, las 2 fuentes principales de AGE sistémicos provienen del metabolismo anormal de la glucosa y de los alimentos. Del total de AGE ingeridos, aproximadamente el 10% es absorbido y solo un tercera parte se excreta. El contenido de AGE en los alimentos puede aumentar de 10-200 veces dependiendo de las condiciones utilizadas para su preparación6.
Estructura del RAGE y sus ligandosEl receptor para productos finales de glicación avanzada (RAGE) es un receptor multiligando perteneciente a la superfamilia de las inmunoglobulinas, que se expresa de manera constitutiva en una amplia gama de células adultas diferenciadas, tales como cardiomiocitos, neuronas, neutrófilos, monocitos/macrófagos, linfocitos, células dendríticas y células endoteliales vasculares, entre otras, y es considerado un receptor de reconocimiento de patrones por su capacidad para detectar estructuras tridimensionales en lugar de secuencias específicas de aminoácidos. Además de los AGE, los ligandos que se han encontrado para ser reconocidos por RAGE son el péptido β-amiloide, las proteínas de alta movilidad del grupo de caja 1 (HMGB1; llamada también anfoterina), proteínas del grupo S100, productos avanzados de oxidación de proteínas, C3a, integrinas-β2 Mac/CD11b, lipopolisacáridos y fosfatidilserina en la superficie de células apoptóticas.
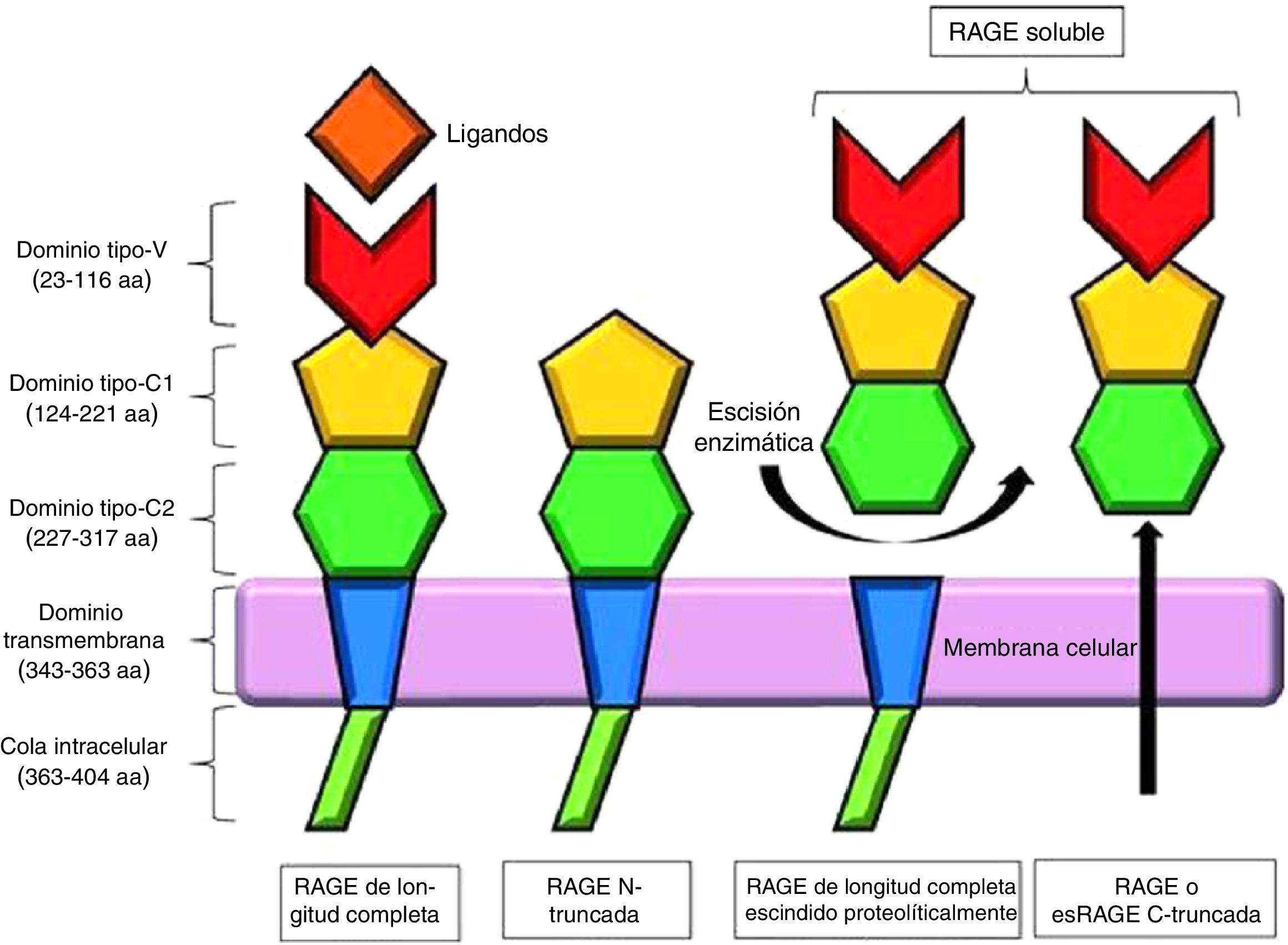
La forma clásica de RAGE, también denominada RAGE de longitud completa, se compone de un único dominio transmembrana, una porción citosólica y una región extracelular. La región extracelular consiste en 2 dominios de inmunoglobulina tipo C (C1 y C2) y un dominio tipo V, considerado como el lugar principal de unión a los ligandos; la eliminación de este dominio da como resultado la forma N-truncada. La porción citosólica es esencial para la señalización intracelular posterior a la unión del ligando en la región V7 (fig. 2).

Representación esquemática de los receptores de los productos finales de glicación avanzada y sus isoformas. Modificada de Chuah et al.7.
Además del RAGE de longitud completa, existen 19 variantes de empalme, las cuales, de acuerdo con el Comité de Nomenclatura de Genes Humanos, han sido resumidas y renombradas en RAGE_v1 a RAGE_v19. Se han identificado 2 formas solubles de RAGE en el plasma humano: RAGE_v1 o esRAGE, que resultan de un splicing alternativo, y sRAGE, una isoforma de escisión proteolítica en la porción extracelular del RAGE de longitud completa, mediada por la metaloproteinasa ADAM 107–9.
Hasta el momento no se cuenta con ensayos clínicos controlados que demuestren la importancia de sRAGE en humanos; sin embargo, diversos estudios han señalado que niveles disminuidos se asocian a una mayor incidencia de eventos cardiovasculares4.
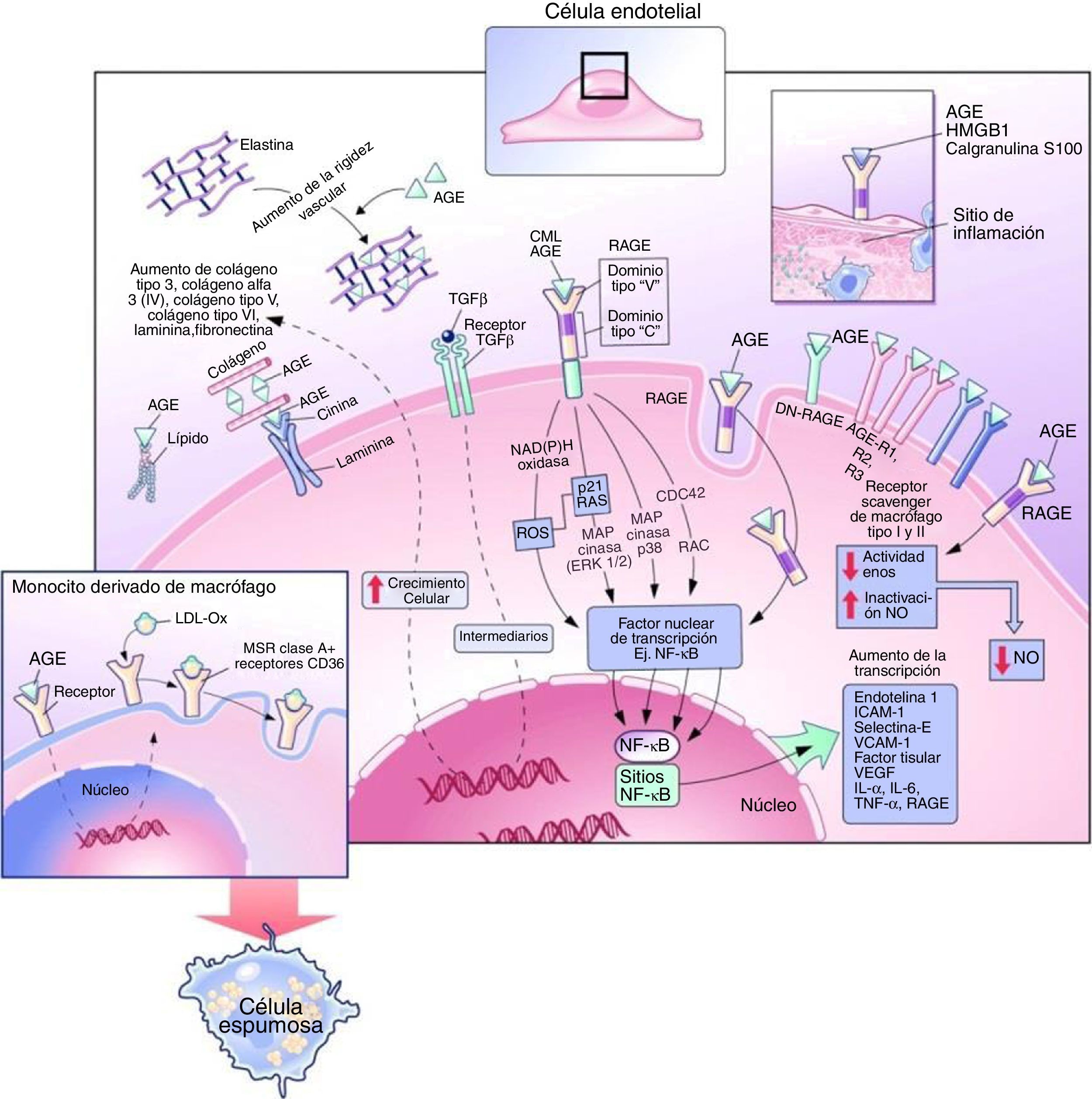
Vía de señalización ligando-RAGELa interacción de RAGE con sus ligandos induce la activación del NF-KB a través de múltiples vías de transducción de señales intracelulares, incluyendo proteínas cinasas activadas por mitógenos (MAPK), cinasas reguladas por signos extracelulares 1/2 (ERK 1/2), p38, la NADPH oxidasa, la cinasa terminal Jun-N, p21/RAS, GTPasas Cdc42 y Rac10.
Tras su activación, el NF-KB pasa al núcleo celular e inicia la transcripción de diversas proteínas que favorecen el desarrollo y la progresión de las complicaciones vasculares, tales como moléculas de adhesión celular vascular-1, factor de crecimiento transformante-β1, factor de crecimiento de tejido conectivo, factor de crecimiento derivado de las plaquetas, factor de necrosis tumoral-α, IL-1 e IL-6. A su vez, el ADN también codifica para la expresión de RAGE, actuando como un mecanismo de retroalimentación positiva4 (fig. 3).

Efectos intracelulares y extracelulares de los productos finales de glicación avanzada. Modificada de Goldin et al.10.
En los fibroblastos, la interacción AGE-RAGE modula la expresión de colágeno; en células musculares lisas modula su migración y proliferación; en fagocitos mononucleares modula la quimiotaxis y la haptotaxis, además de la expresión de moléculas proinflamatorias y protrombóticas; y en los linfocitos estimula la generación de IL-211.
Estrés oxidativo y disfunción endotelialNumerosos estudios han informado de que el estrés oxidativo se incrementa en pacientes diabéticos, jugando un papel importante en la patogénesis de las complicaciones vasculares12,13.
El mecanismo principal por el cual la interacción AGE-RAGE genera estrés oxidativo es mediante la activación de la NADPH oxidasa, provocando un aumento de especies reactivas de oxígeno, llevando así a la oxidación de lípidos, proteínas y ADN13,14. Un factor adicional que contribuye en este proceso es la glicación de antioxidantes como la Cu/Zn superóxido dismutasa4.
Además de la interacción AGE-RAGE, otras vías implicadas en el estrés oxidativo son la de los polioles, la de la hexosamina y la de la proteína cinasa C. Es interesante conocer que los AGE también se forman durante el envejecimiento, y que el envejecimiento vascular también se caracteriza por la activación de NF-KB, el aumento de factor de necrosis tumoral-α, la NADPH oxidasa y la disfunción endotelial15.
Se cree que la hiperglucemia afecta la función endotelial reduciendo la producción de óxido nítrico e incrementando la síntesis de vasoconstrictores potentes como la endotelina y la angiotensina ii(Ang II)16,17 (fig. 3). En pacientes con diabetes tipo 2, los niveles de AGE en suero son inversamente proporcionales al grado de vasodilatación endotelial. Un mecanismo evidencia que los AGE reducen la vida media de la sintasa de óxido nítrico endotelial a través del aumento de la tasa de degradación del ARNm. Otro mecanismo propone que los AGE alteran la producción de óxido nítrico a través de la unión de residuos de carboximetil-lisina a receptores de AGE en el endotelio, causando una reducción en la fosforilación de residuos de serina en sintasa de óxido nítrico endotelial, lo que resulta en la desactivación de la enzima10.
Los mecanismos precisos que conducen a la hiperpermeabilidad vascular observada en la vasculopatía diabética siguen siendo poco claros, sin embargo; se ha demostrado que los AGE pueden inducir y perturbar la barrera fisiológica del endotelio mediante la vía de señalización RAGE/Rho18. Los cambios observados por activación de esta vía son pérdida o debilitamiento en las uniones adherentes, redistribución de las uniones focales, reacomodamiento del citoesqueleto y contracción celular.
Vía de la quimasaCerca del 70% de los pacientes con diabetes tienen también hipertensión, lo que amplifica o acelera el desarrollo de las complicaciones micro y macrovasculares, además de contribuir a la disfunción endotelial y a la remodelación vascular, incluyendo el aumento del espesor de la pared vascular, la hiperplasia de las células del músculo liso vascular y la acumulación de matriz extracelular en arterias de resistencia.
La Ang II es un mediador clave en la enfermedad vascular diabética. A pesar de que la enzima convertidora de angiotensina se relaciona como la principal productora predominante de Ang II, la evidencia actual muestra otras vías alternas para su producción, como la de la quimasa.
Esta vía es responsable de hasta el 80% de la producción local de Ang II en el corazón y en las arterias coronarias. Su expresión está más caracterizada en los mastocitos, aunque también se ha observado su expresión en células del músculo liso vascular, en el mesangio glomerular y en células epiteliales. Los AGE son capaces de inducir la expresión de la quimasa vascular a través de la vía de señalización RAGE-ERK1/1 MAPK19.
Complicaciones vasculares de la diabetesMiocardiopatía diabéticaEn el corazón, el 70-80% de la masa celular está compuesta por miocitos; el 20-30% restante incluye células musculares lisas vasculares, células endoteliales y mayoritariamente fibroblastos. Estos últimos son responsables del mantenimiento homeostático y de las alteraciones patológicas de la matriz extracelular (MEC) observadas en el corazón20.
En condiciones de hiperglucemia observadas en pacientes diabéticos, los estímulos bioquímicos y mecánicos alteran la comunicación entre los fibroblastos y la MEC, conduciendo a un aumento de la rigidez cardiaca e impactando negativamente en su función20,21.
La activación de p38, ERK 1/2, NF-KB y cinasa terminal Jun-N por medio de la interacción AGE-RAGE ha demostrado movilizar múltiples factores de transcripción que estimulan la expresión de factores de crecimiento y la acumulación de proteínas de la MEC. El factor de crecimiento transformante-β, muy probablemente regulado en un punto intermedio en estas vías, promueve la síntesis de colágeno tipo iii, iv, v y vi, laminina y fibronectina10,20.
Rap1A, un miembro de la familia RAS, ha demostrado participar en vías hipertróficas. Bajo condiciones de hiperglucemia, Rap1A aumenta la actividad de ERK 1/2, y en última instancia es capaz de influir en otras vías de señalización, incluyendo AGE-RAGE20.
Un efecto adicional de los AGE es su capacidad de alterar las propiedades del colágeno, la vitronectina y la laminina a través de enlaces covalentes intermoleculares o por entrecruzamiento, cuyo efecto parece inhibir la degradación de la MEC10,21.
AterosclerosisLos factores que subyacen a la aterosclerosis acelerada en la diabetes se extienden más allá de la dislipidemia, la hipertensión y la obesidad. Diversos estudios evidencian que el inicio de la aterosclerosis está estrechamente ligado a la disfunción endotelial22.
La activación del NF-KB a través del eje AGE-RAGE en las células vasculares conduce a la expresión de múltiples moléculas proinflamatorias y moléculas de adhesión (por ejemplo, moléculas de adhesión celular vascular-1), facilitando la adhesión de monocitos, seguida de su migración hacia el espacio subendotelial y su diferenciación en macrófagos1.
Los AGE contribuyen a la expresión de receptores de LDL oxidadas en los macrófagos, incluyendo receptores scavenger, CD36 y el receptor LDL oxidasa tipo lectina 1. El aumento de expresión de estos receptores y la glicación de la apolipoproteína B en LDL mejora la captación de LDL oxidasa, lo que resulta en una mayor transformación a células espumosas7,8,10 (fig. 3).
La acumulación de células espumosas en el espacio subendotelial da origen a estrías grasas en la pared del vaso; estas lesiones continúan desarrollándose, caracterizándose por la acumulación de células musculares lisas, la formación de un núcleo necrótico y la acumulación de lípidos, así como la formación de una capa fibrosa1.
Aunque la aterosclerosis se inicia por la deposición de lipoproteínas ricas en colesterol en la pared arterial, la entrada de leucocitos inflamatorios aumenta el riesgo de enfermedad arterial crónica. Una posible explicación a este hecho es la mayor producción de S100A8/A9 por los neutrófilos como respuesta a la hiperglucemia. Después de su síntesis, S100A8/A9 puede unirse a RAGE y estimular directamente la proliferación de polimorfonucleares o alternativamente conducir a la producción de citocinas proliferativas que actúen en otras células progenitoras mieloides23.
Algunas de estas lesiones avanzadas se vuelven inestables con la posibilidad de romperse, lo que desencadena los eventos tromboembólicos, que dan lugar a las manifestaciones clínicas de las enfermedades cardiovasculares como el infarto de miocardio y el accidente cerebrovascular.
La interacción AGE-RAGE induce un estado procoagulante en las células endoteliales que resulta de la reducción de la actividad de la trombomodulina en paralelo con el aumento de expresión del factor tisular1.
RetinopatíaLa retinopatía se ha convertido en la complicación vascular más común asociada con la diabetes y la principal causa de ceguera entre los adultos de 20 a 74 años. El mal control de la glucemia y la presión arterial complican esta enfermedad1,24.
El Diabetes Control and Complications Trial y el UK Prospective Diabetes Study confirman la importancia de un buen control de la glucemia para retrasar el desarrollo y la progresión de la retinopatía diabética. A pesar de ello, esta enfermedad se produce incluso con un buen control de la glucemia12,24.
Múltiples vías se han asociado a la patogénesis de la retinopatía diabética, incluyendo la vía de los polioles, la activación de diacilglicerol y la vía de la proteína cinasa C; no obstante, la interacción AGE-RAGE ha sido investigada más recientemente1.
Clínicamente esta afección se clasifica en retinopatía diabética no proliferativa y proliferativa. La primera se caracteriza por microangiopatía capilar, microaneurismas, engrosamiento de la membrana basal y pérdida de pericitos. Muchos de estos eventos son a consecuencia de la disfunción endotelial producida por la interacción AGE-RAGE. En la retinopatía diabética proliferativa la activación de RAGE en la glía de Müller resulta en la activación de ERK 1/2 y la producción posterior de citocinas inflamatorias como el VEGF y la MCP-1, lo que implica un papel crítico de RAGE en la neovascularización y la selección de células inmunes en las capas de la retina1,8,12.
Otras consecuencias de los efectos nocivos de RAGE en la retinopatía diabética incluyen la rotura de la barrera hematorretiniana y el aumento de leucotaxis1.
NefropatíaLa nefropatía diabética, que se caracteriza por el desarrollo de proteinuria y una disminución de la tasa de filtración glomerular, representa la principal causa de enfermedad renal terminal en el mundo occidental.
Un aspecto de gran importancia es la acumulación de AGE que ocurre a nivel renal en pacientes diabéticos1,14. En este órgano, RAGE se expresa en los podocitos y en las células endoteliales. Uno de los efectos producidos por los AGE a nivel renal es la hipofunción de la glioxalasa i, una enzima citosólica encargada de desintoxicar el MG. Por tanto, el riñón contribuye a mayores niveles de AGE en plasma1,11.
Estudios en ratones transgénicos que sobreexpresan RAGE mostraron aumentos significativos en el peso del riñón, albuminuria y glomeruloesclerosis, poniendo de manifiesto una vez más la importancia de RAGE8.
NeuropatíaLa neuropatía diabética se caracteriza por una alteración en la sensibilidad a las vibraciones, a los umbrales térmicos y al dolor. Las lesiones endoteliales ocasionadas por los AGE pueden afectar el flujo de sangre, conducir a un estado de hipoxia, estrés oxidativo y, finalmente, al deterioro de la fibra nerviosa1,8.
La interacción AGE-RAGE y la progresión de esta enfermedad se han demostrado por la identificación de RAGE tanto en vasos endoneurales como perineurales.
Además, algunos estudios en modelos experimentales han demostrado que HMGB1, un ligando de RAGE, contribuye al dolor neuropático después de una lesión nerviosa, y la interrupción de la señalización HMGB1/RAGE podría ser una estrategia terapéutica prometedora1.
Tratamientos y avances terapéuticosLa mayoría de los tratamientos actuales para reducir el riesgo cardiovascular relacionado con la diabetes mellitus van dirigidos hacia el buen control de la hiperglucemia, la hipertensión arterial, los niveles de ácidos grasos libres, el peso corporal e, indiscutiblemente, cambios en el estilo de vida16. Actualmente numerosos compuestos que previenen las complicaciones inducidas por AGE están bajo investigación; sin embargo, aún se encuentran en etapas experimentales.
La aminoguanidina, un compuesto utilizado en modelos experimentales, afecta favorablemente la estructura vascular, aumenta la elasticidad arterial, reduce la acumulación de MEC y de AGE, así como la gravedad de la placa aterosclerótica. En un ensayo controlado aleatorizado con placebo en pacientes con diabetes mellitus tipo 1, la aminoguanidina redujo la proteinuria y la progresión de la retinopatía10,11.
ALT-946 y ALT-711 son otros compuestos en etapa experimental. Experimentos en ratas tratadas con ALT-711 mostraron un aumento de la solubilidad del colágeno con una disminución en la expresión de RAGE, además de observarse mejorías en la función cardiaca10.
Otros experimentos en modelos animales han demostrado que la piridoxamina reduce los niveles plasmáticos de AGE y la hiperlipidemia10.
ConclusionesLos AGE son un grupo heterogéneo de compuestos que toman gran relevancia en el inicio y el desarrollo de las complicaciones vasculares en pacientes diabéticos; no solo su formación acelerada como consecuencia de la hiperglucemia sostenida conlleva la aparición de dichas alteraciones, pues existen otras vías alternas, como la de los polioles, la de la hexosamina y la de la proteína cinasa C, que potencian el daño celular.
El estrés oxidativo y la disfunción endotelial inducida por las diversas vías de señalización mediadas por el eje ligando-RAGE son, hasta ahora, los principales factores que deterioran la función celular y, por tanto, la homeostasis de diversos sistemas fisiológicos.
A pesar de los avances significativos en posibles tratamientos, el conocimiento detallado de las vías de señalización y de los diferentes niveles de regulación mediados por RAGE abre varias posibilidades para futuras intervenciones terapéuticas.
FinanciaciónNo se recibió patrocinio para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.