




A síndrome de Cushing cíclica (SCC) é rara e caracteriza‐se por episódios de hipercortisolismo, intercalados com períodos de secreção normal de cortisol. É diagnosticada pela identificação de 3 picos e 2 vales na produção de cortisol. Os autores descrevem um caso de SCC, alertando para os seus desafios e particularidades, e realizam uma revisão da literatura.
Doente do sexo feminino, 32 anos, com clínica de hipercortisolismo e avaliação analítica compatível com síndrome de Cushing. No estudo etiológico obteve‐se hormona adrenocorticotrófica (ACTH) doseável (45pg/mL), frenação <50% do cortisol na prova de dexametasona 8mg (44%) e resposta excessiva de ACTH (96%) e cortisol (63%) na prova de CRH. A ressonância magnética nuclear selar foi sugestiva de microadenoma hipofisário. Para confirmacão de doença de Cushing foi programado cateterismo dos seios petrosos inferiores, sem confirmação prévia de hipercortisolismo. Obteve‐se um gradiente de ACTH central/periférico pós CRH < 3, inconclusivo para causa hipofisária. Os exames de localização de tumor ectópico foram negativos. Dada a incerteza diagnóstica, optou‐se inicialmente por vigilância laboratorial, identificando‐se mais de 3 picos e 2 vales na produção de cortisol, sugerindo o diagnóstico de SCC. A clínica foi flutuante, coincidindo com os períodos de hipercortisolismo. Decidiu‐se então por cirurgia transesfenoidal e o exame histológico foi compatível com adenoma hipofisário positivo para ACTH. Constatou‐se remissão da doença.
Os autores alertam para a importância de requisitar doseamentos laboratoriais frequentes quando se suspeita de SCC, com o objetivo de identificar picos e vales na produção de cortisol. No momento de realização do cateterismo, a doente provavelmente estaria numa fase de produção normal de cortisol, o que condicionou o resultado inconclusivo e o atraso no diagnóstico e terapêutica. O estudo etiológico deve ser efetuado durante uma fase de excesso de cortisol, exigindo confirmação analítica prévia.
Cyclic Cushing's syndrome (CCS) is a rare disorder, characterized by episodes of hypercortisolism, interspersed by periods of normal cortisol secretion. Its diagnosis is made by identifying 3 peaks and 2 troughs of cortisol production. The authors report a case of CCS, addressing its challenges and features, and perform a review of the literature.
A 32‐year‐old woman presented with clinical signs of hypercortisolism and biochemical evaluation was consistent with Cushing's syndrome. Tests to define its cause revealed a detectable adrenocorticotropic hormone (ACTH) (45pg/mL), <50% cortisol decrease following high dose dexamethasone suppression test (44%) and exaggerated response of ACTH (96%) and cortisol (63%) after CRH stimulation test. Pituitary magnetic resonance imaging identified a pituitary microadenoma. To confirm the diagnosis of Cushing's disease, an inferior petrosal sinus sampling was scheduled without prior confirmation of hypercortisolism. An ACTH petrosal sinus/peripheral ratio < 3 after CRH administration was found, inconclusive for a pituitary origin. Exams to localize an ectopic tumor were negative. Owing to the diagnostic uncertainty, the authors initially decided to monitor the patient and identified more than 3 peaks and 2 troughs of cortisol production, suggesting the diagnosis of CCS. Clinical signs were fluctuating and appeared during periods of hypercortisolism. We then decided to perform a transsphenoidal surgery and histological examination revealed an ACTH positive pituitary adenoma. Disease remission was achieved.
The authors highlight the need of frequent laboratory measurements when CCS is suspected, to identify peaks and troughs of cortisol production. When sampling was performed, the patient would probably be in a period of normal cortisol production, contributing to the inconclusive result and the delay in proper diagnosis and therapy. Tests used to determine the cause of CCS should be carried out during a period of cortisol excess, requiring prior confirmation of hypercortisolism.
A síndrome de Cushing (SC) resulta da exposição prolongada dos tecidos a níveis inapropriadamente elevados de glicocorticóides. A causa mais frequente é a administração exógena de substâncias contendo glicocorticóides. O hipercortisolismo endógeno é raro (incidência anual 2‐3 casos por milhão de habitantes)1,2 e apresenta várias etiologias, nomeadamente hipofisária (doença de Cushing), suprarrenal e tumor ectópico. A clínica é variada e pouco específica, salientando‐se: estrias purpúreas, pletora facial, equimoses fáceis, miopatia proximal, aumento ponderal, irregularidades menstruais, hirsutismo, hipertensão arterial, entre outros. Quando há suspeita de SC, deve ser efetuada uma marcha diagnóstica com o objetivo de identificar a síndrome e definir a sua causa. A terapêutica adequada permite reduzir a morbilidade e a mortalidade associadas3,4.
A síndrome de Cushing cíclica (SCC) é uma variante rara da SC, caracterizada por episódios de hipercortisolismo, intercalados com períodos de secreção normal de cortisol5. O seu diagnóstico é complexo e exige doseamentos laboratoriais frequentes, sendo efetuado pela identificação de 3 picos e 2 vales na produção de cortisol5,6.
Os autores descrevem um caso de SCC, alertando para os desafios e particularidades deste diagnóstico, e realizam uma revisão da literatura.
Caso clínicoDoente do sexo feminino, 32 anos, caucasiana, secretária, com um filho (6 anos), referenciada à consulta de Endocrinologia do Hospital Garcia de Orta, em novembro/2000, por quadro clínico com 7 anos de evolução, caracterizado por aumento ponderal de 16kg (57 para 73 kg), distribuição centrípeta da gordura corporal e equimoses fáceis. Negava aumento da pilosidade, acne, irregularidades menstruais, astenia, mialgias ou outra sintomatologia associada. Sem antecedentes pessoais e familiares valorizáveis. Não fazia qualquer tipo de medicação. Ao exame objetivo: peso 73kg; altura 168cm; índice de massa corporal 25,9kg/m2; pressão arterial 127/64mmHg; frequência cardíaca 85/minuto. Clínica sugestiva de hipercortisolismo, nomeadamente pletora facial e face em lua‐cheia; sem estrias purpúreas, equimoses, hirsutismo, nem outras alterações relevantes (fig. 1).
.")
Analiticamente, destacava‐se aumento do cortisol livre urinário (CLU) de 24 horas em 2 doseamentos (147,5 e 115,5μg [15‐90]), ausência de ritmo circadiano de cortisol sérico (matinal 14,2μg/dL [5‐30], noturno 13,9μg/dL [<7,5]), bem como ausência de frenação adequada do cortisol sérico nas provas de dexametasona 1mg, 2mg e 2mg com CRH, 8,1μg/dL [<1,8], 9,5μg/dL [<1,4] e 14,2μg/dL [< 1,4], respetivamente.
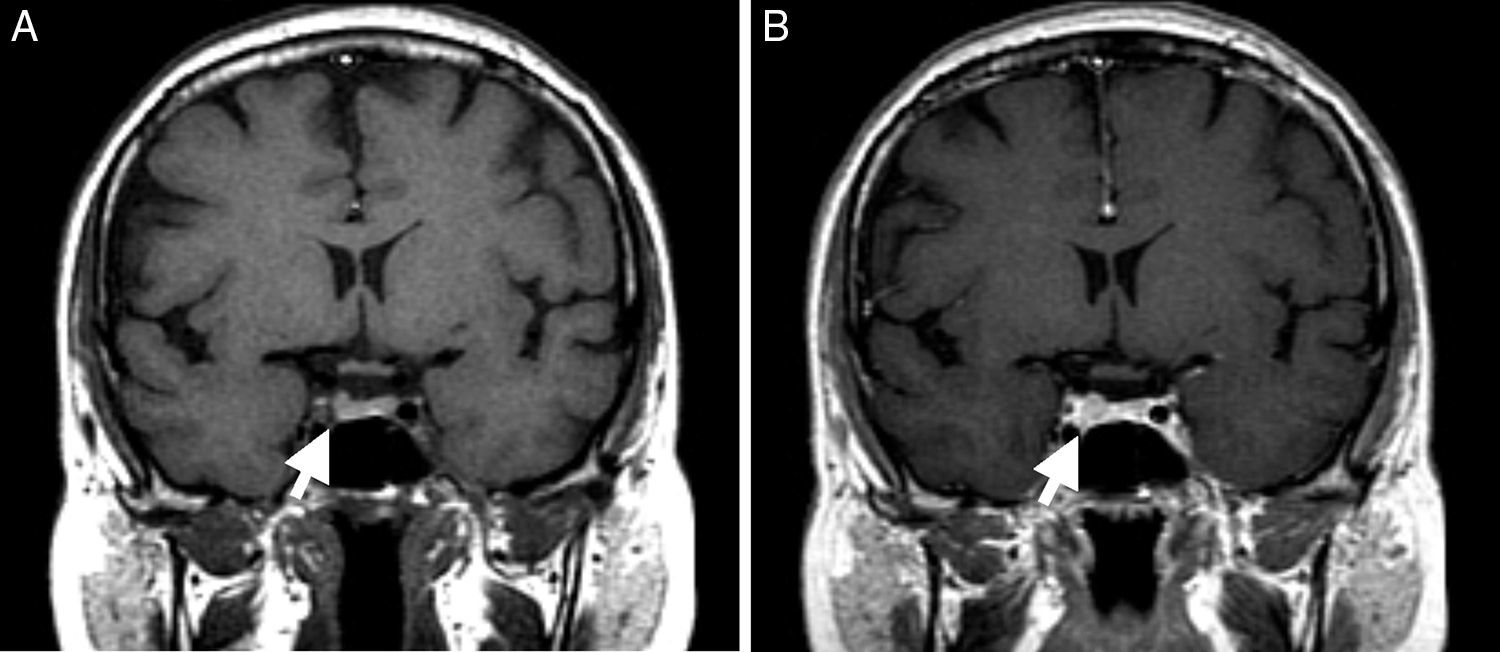
Estes aspetos confirmaram SC, procedendo‐se subsequentemente à identificação da sua etiologia. Obteve‐se hormona adrenocorticotrófica (ACTH) doseável de 45pg/mL, frenação <50% do cortisol sérico na prova de dexametasona 8mg (basal 15,8μg/dL, final 8,8μg/dL, frenação 44%) e resposta excessiva de ACTH e cortisol na prova de CRH (ACTH basal 47,7pg/mL, pico de ACTH aos 15 minutos 93,7pg/mL, correspondendo a aumento de 96%; cortisol basal 14,1μg/dL, pico de cortisol aos 30 minutos 23μg/dL, traduzindo‐se em aumento de 63%). Foi realizada ressonância magnética nuclear (RMN) selar, sugestiva de microadenoma hipofisário lateral direito com elevação do bordo hipofisário, sem alteração do sinal antes e após contraste (fig. 2). Restante função hipofisária e androgénios sem alterações.
 e com gadolínio (B). Identifica‐se microadenoma na hemi‐hipófise direita (seta), com elevação do bordo hipofisário, sem alteração do sinal antes e após contraste.")
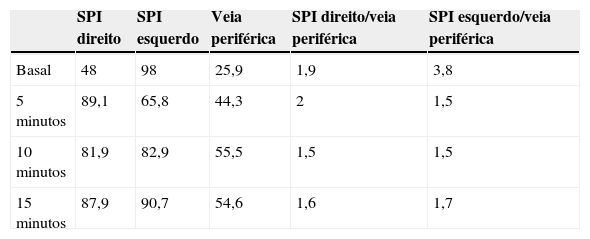
Embora o quadro fosse sugestivo de doença de Cushing, a supressão <50% na prova de dexametasona 8mg levou à realização de cateterismo dos seios petrosos inferiores em fevereiro/2002, sem confirmação prévia de hipercortisolismo. Apesar do gradiente de ACTH central esquerdo/periférico basal ser >2, o gradiente pós CRH <3 levou os autores a questionarem o diagnóstico de doença de Cushing (tabela 1). O Octreoscan e a tomografia computadorizada (TC) torácica foram negativos para tumor ectópico e a TC das glândulas suprarrenais identificou discreto esboço nodular bilateral.
Doseamentos de ACTH (pg/mL) no cateterismo dos seios petrosos inferiores com CRH. Apesar do gradiente de ACTH central esquerdo/periférico basal ser >2, obteve‐se um gradiente pós CRH <3. SPI: seio petroso inferior
| SPI direito | SPI esquerdo | Veia periférica | SPI direito/veia periférica | SPI esquerdo/veia periférica | |
|---|---|---|---|---|---|
| Basal | 48 | 98 | 25,9 | 1,9 | 3,8 |
| 5 minutos | 89,1 | 65,8 | 44,3 | 2 | 1,5 |
| 10 minutos | 81,9 | 82,9 | 55,5 | 1,5 | 1,5 |
| 15 minutos | 87,9 | 90,7 | 54,6 | 1,6 | 1,7 |
ACTH: hormona adrenocorticotrófica; CRH: hormona libertadora de corticotropina.
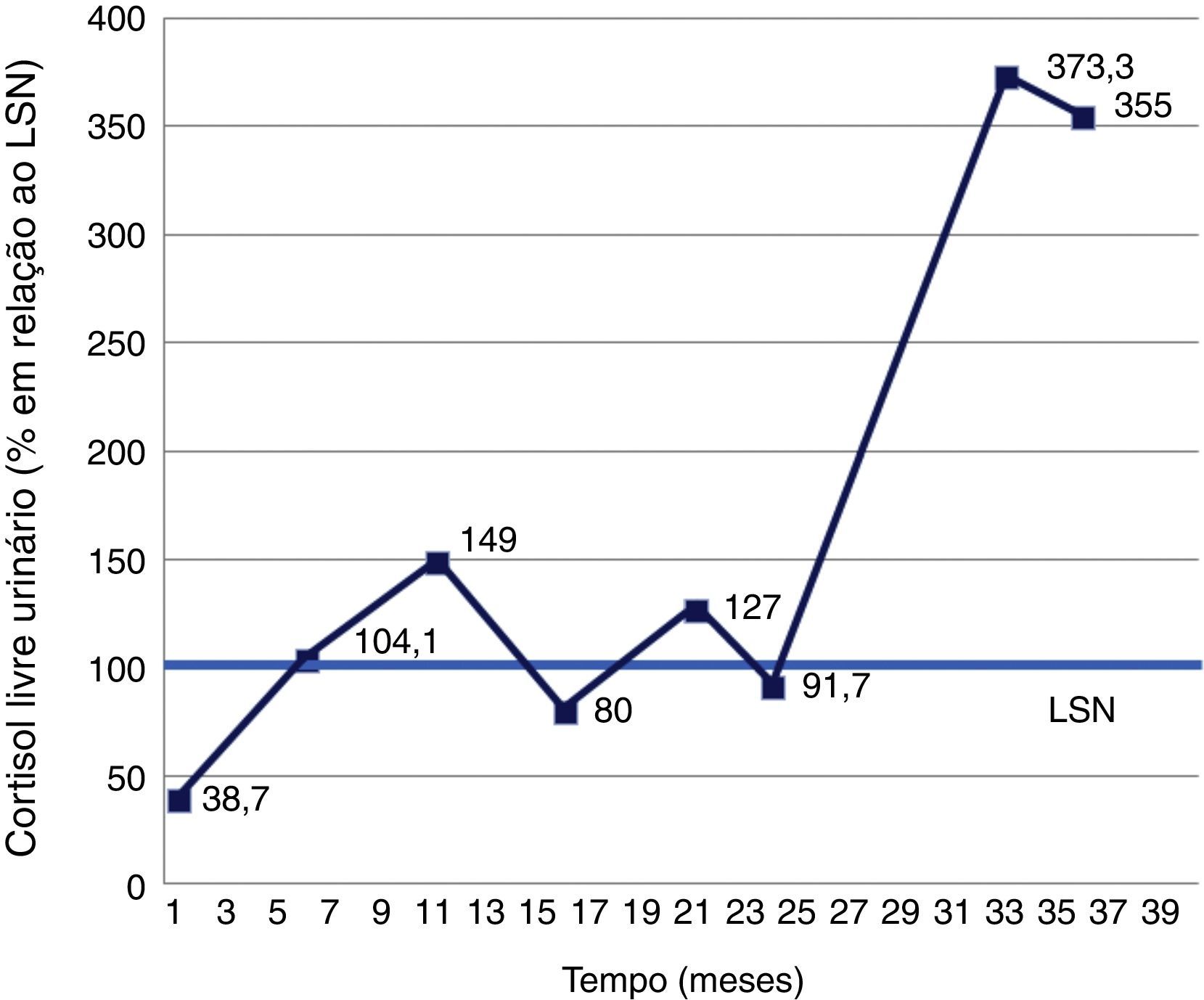
Dada a dúvida etiológica, optou‐se inicialmente por vigilância clínica e analítica, com monitorização do CLU. Entre julho/2002 e setembro/2005 ocorreram períodos de hipercortisolismo bioquímico, alternando com valores dentro da normalidade. Foi possível a identificação de 3 picos e 2 vales, sugerindo o diagnóstico de SCC (fig. 3). A presença de sintomas e sinais da SC, nomeadamente aumento ponderal, pletora facial e face em lua‐cheia, foi flutuante, coincidindo com os períodos de elevação do cortisol (fig. 4).
 e setembro/2005 (mês 39). Uma vez que os resultados foram obtidos de laboratórios diferentes, optou‐se por apresentar o valor de cortisol em relação ao respetivo limite superior de normalidade (LSN). Observaram‐se períodos de hipercortisolismo, alternando com valores normais. Foi possível a identificação de 3 picos e 2 vales, sugerindo o diagnóstico de síndrome de Cushing cíclica.")
Doseamentos de cortisol livre urinário de 24 horas, entre julho/2002 (mês um) e setembro/2005 (mês 39). Uma vez que os resultados foram obtidos de laboratórios diferentes, optou‐se por apresentar o valor de cortisol em relação ao respetivo limite superior de normalidade (LSN). Observaram‐se períodos de hipercortisolismo, alternando com valores normais. Foi possível a identificação de 3 picos e 2 vales, sugerindo o diagnóstico de síndrome de Cushing cíclica.
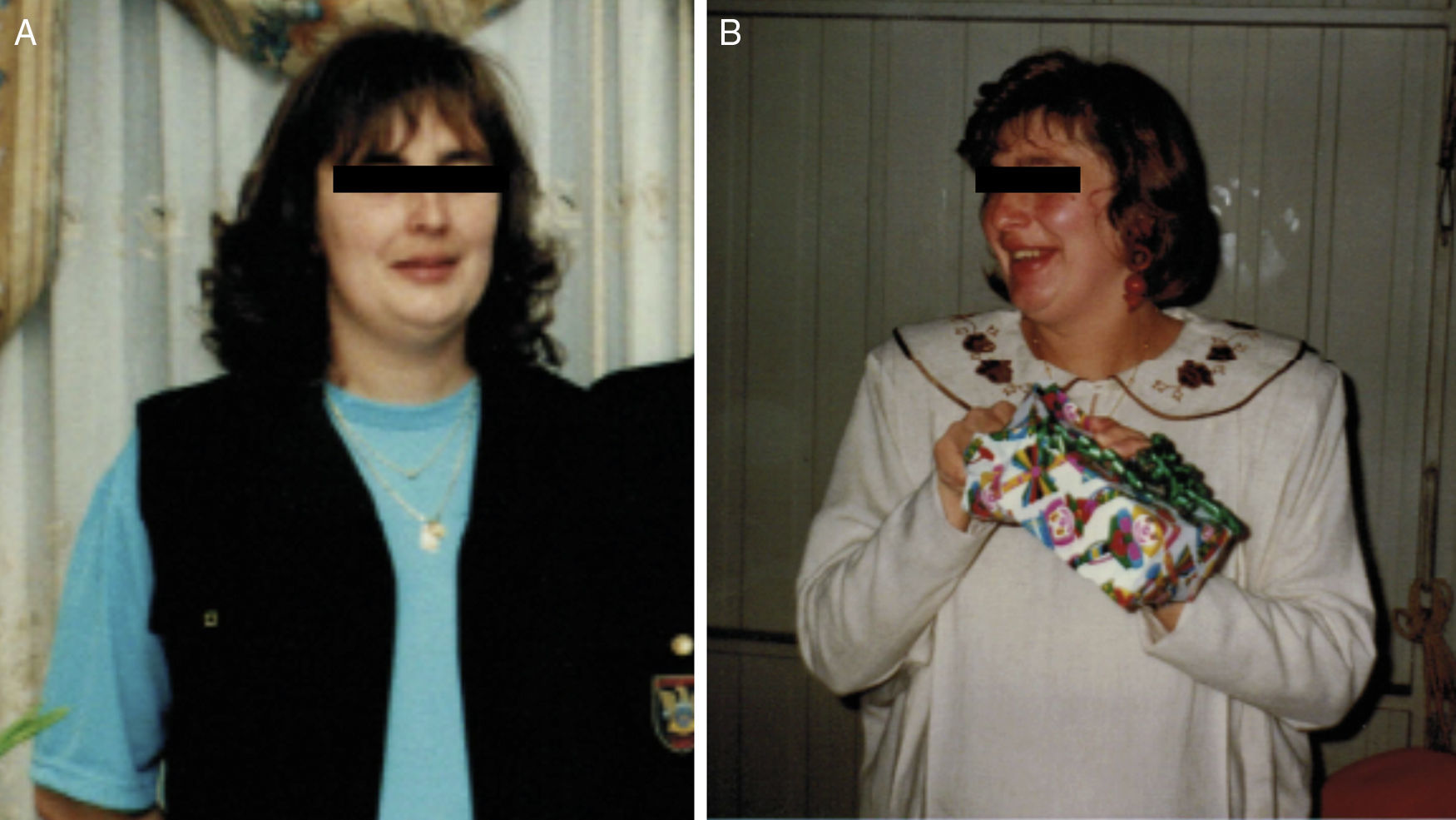
 Ausência de clínica associada à síndrome de Cushing, correspondendo a um período de secreção normal de cortisol (julho/2002). B) Presença de algumas das características da síndrome, nomeadamente aumento ponderal, pletora facial e face em lua‐cheia, em fase de hipercortisolismo laboratorial (maio/2003).")
Fotografias da doente durante o seguimento. A) Ausência de clínica associada à síndrome de Cushing, correspondendo a um período de secreção normal de cortisol (julho/2002). B) Presença de algumas das características da síndrome, nomeadamente aumento ponderal, pletora facial e face em lua‐cheia, em fase de hipercortisolismo laboratorial (maio/2003).
Decidiu‐se por cirurgia por via transesfenoidal do adenoma da hemi‐hipófise direita, efetuada em outubro/2005, sem complicações no pós‐operatório. O exame histológico foi compatível com adenoma hipofisário com marcação imuno‐histoquímica para ACTH e negativo para hormona de crescimento, prolactina, hormona folículo‐estimulante, hormona luteinizante e hormona estimuladora da tiróide. A doente foi medicada com hidrocortisona 15mg/dia, com redução progressiva da dose, suspendendo corticoterapia 16 meses após a intervenção cirúrgica (fevereiro/2007).
Desde então observou‐se regressão progressiva do quadro clínico com perda ponderal de 20kg e ausência de estigmas de hipercortisolismo (fig. 5). Apresenta‐se em remissão da doença de Cushing, com valores normais de CLU de 24 horas (último doseamento 24μg/dL [10‐60]), ritmo circadiano de cortisol mantido, função hipofisária basal preservada e RMN selar sem evidência de adenoma.
Discussão. Perda ponderal de 20kg e desaparecimento dos estigmas de hipercortisolismo.")
A SCC foi descrita pela primeira vez por Bailey em 1971, numa doente com SC ectópico7. Desde então outros casos foram publicados e uma revisão de 2007 identificou 65 casos na literatura5.
Encontram‐se várias designações desta entidade, nomeadamente cíclica, periódica, intermitente, flutuante, variável, episódica e imprevisível5,8. Os episódios de hipercortisolismo clínico e/ou bioquímico podem ser regulares ou irregulares, sendo intercalados por períodos, com duração variável, de secreção normal de cortisol ou mesmo de hipocortisolismo5.
A fisiopatologia da SCC é desconhecida. Alguns autores defendem que o hipercortisolismo intermitente está associado a episódios espontâneos de necrose/hemorragia tumoral9–11. Outras hipóteses foram propostas, nomeadamente a existência de feedback negativo do cortisol sob a ACTH tumoral12, bem como a possível variabilidade periódica de estímulos hipotalâmicos condicionando maior ou menor secreção de ACTH5,13. No entanto, o mecanismo exato continua por definir14.
A clínica é variada, podendo o doente apresentar‐se com sinais e sintomas constantes ou flutuantes coincidindo com os episódios de hipercortisolismo laboratorial. Numa minoria de casos não há evidência da clínica sugestiva de SC5,8.
Shapiro et al. classificaram os doentes com SCC em 4 categorias de acordo com o padrão de hipersecreção de cortisol (regular ou irregular) e a presença ou ausência de flutuações concomitantes do quadro clínico15.
A avaliação do SCC exige doseamentos laboratoriais frequentes. São preferencialmente utilizados o CLU de 24 horas e/ou o cortisol salivar noturno, com identificação de valores elevados (picos), alternando com resultados normais ou baixos (vales)3,5,16. As provas de dexametasona não estão recomendadas dado poderem originar uma resposta paradoxal de cortisol6,17. Um estudo recente sugere que o doseamento de cortisol no cabelo pode vir a ser uma ferramenta útil no diagnóstico de SCC18.
Deve suspeitar‐se desta entidade em doentes com sinais/sintomas típicos, mas com resultados analíticos normais ou discrepantes, bem como em casos assintomáticos com dados laboratoriais sugestivos de excesso de cortisol5. O diagnóstico diferencial inclui quadros ligeiros de SC, SC subclínica, pseudo‐Cushing, SC mediada por recetores aberrantes, SC factícia e resistência aos glicocorticóides5,19.
Os critérios de diagnóstico de SCC atualmente aceites incluem a identificação de 3 picos e 2 vales na produção de cortisol5,6. No entanto, esta variabilidade pode ser difícil de identificar, especialmente em doentes com vales de cortisol muito longos, condicionando um atraso no diagnóstico e consequente aumento da morbilidade e mortalidade associadas à SC5,8. Daí a importância de repetir a avaliação laboratorial ao longo do tempo em doentes com suspeita de SCC. Sempre que possível deve proceder‐se à determinação analítica durante períodos de hipercortisolismo clínico3.
Velez et al. sugeriram um protocolo de rastreio de SCC baseado na realização de 4 doseamentos dos seguintes parâmetros: CLU de 24 horas, cortisol salivar noturno, cortisol sérico e ACTH durante períodos de hipercortisolismo clínico. Resultados negativos nas 4 determinações excluem a presença de SCC, exceto quando a suspeição permanece elevada. Resultados intermitentemente positivos podem sugerir a presença de SCC, justificando avaliação adicional20.
A SCC tem sido referida como rara, mas acredita‐se que a dificuldade no diagnóstico possa levar a que seja subdiagnosticada. Alguns estudos avaliaram doentes com SC e demonstraram a presença de produção cíclica de cortisol em 15‐40%5,8.
As provas e os exames efetuados na determinação da etiologia são semelhantes à SC em geral. Uma particularidade da SCC está relacionada com a obrigatoriedade de confirmar hipercortisolismo, por exemplo com CLU, antes do estudo etiológico5,8,19. No caso da doente descrita não foi previamente confirmado hiperticortisolismo e provavelmente estaria numa fase de produção normal de cortisol na altura do cateterismo, o que condicionou o resultado inconclusivo do exame e o atraso no correto diagnóstico e sua terapêutica.
A SCC na doente só foi confirmada posteriormente, durante o período de follow‐up que se seguiu à indefinição etiológica. Foram identificados mais do que 3 picos e 2 vales de CLU, com manifestação das características clínicas típicas da SC nas fases de produção aumentada de cortisol. A decisão pela cirurgia transfenoidal foi acertada, uma vez que o resultado histológico confirmou doença de Cushing e a doente se encontra em remissão. Além da remoção tumoral completa e normalização analítica, ocorreu franca melhoria clínica.
Meinardi et al.5 reviram as características dos 65 doentes com SCC descritos na literatura. Observou‐se uma predominância do sexo feminino (3:1), manifestando‐se mais frequentemente na 5.a década de vida. A maioria dos doentes (94%) apresentava pelo menos 2 sinais/sintomas típicos da SC. Doença de Cushing foi identificada em 35 casos (54%), tumor ectópico em 17 casos (26%), causa suprarrenal em 7 casos (11%), sendo a etiologia desconhecida em 6 casos (9%); estes aspetos diferem da SC em geral pela maior prevalência de tumor ectópico na variante cíclica, principalmente tumores carcinóides.
Alexandraki et al.8 identificaram um padrão cíclico em 30 (15%) de 201 indivíduos com doença de Cushing e procederam à comparação das características entre os doentes classificados como cíclicos e não‐cíclicos. Embora nenhum factor fosse específico de doença cíclica, foi encontrada uma associação entre este subgrupo de doentes e uma maior frequência do sexo feminino, idade mais avançada, maior período de follow‐up e menor taxa de identificação histológica do adenoma.
O tratamento da SCC é semelhante à da SC não‐cíclica e depende da etiologia. Alguns estudos indicam que os indivíduos com doença de Cushing cíclica, submetidos a intervenção neurocirúrgica, apresentam taxas de remissão mais baixas e taxas de recorrência mais elevadas do que aqueles com doença não‐cíclica21. No entanto, esta diferença não parece ter significado estatístico e não está provado que o padrão cíclico de hipercortisolismo condicione o prognóstico5,8. Embora alguns autores refiram remissão da doença com terapêutica médica, nomeadamente bromocriptina21–23 e valproato de sódio13, a cirurgia transfenoidal continua a ser a terapêutica de eleição na maioria destes casos. Outro dos aspetos a que devemos estar atentos é que a redução bioquímica após terapêutica pode não refletir remissão, mas sim corresponder a um período de secreção normal de cortisol8,19. Os doentes devem ser seguidos regularmente por longos períodos de tempo para detetar precocemente eventuais recidivas.
Em conclusão, apresenta‐se uma doente com SCC causada por adenoma hipofisário secretor de ACTH. Em caso de suspeição, alerta‐se para a importância de requisitar doseamentos laboratoriais frequentes com o objetivo de identificar picos e vales na produção de cortisol. O estudo etiológico de SCC deve ser efetuado durante uma fase de excesso de cortisol, exigindo confirmação analítica prévia. O diagnóstico e a terapêutica precoces são fundamentais e proporcionam remissão da doença na maioria dos casos, diminuindo a morbilidade e mortalidade associadas à SC.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
Os autores receberam uma bolsa da Sociedade Portuguesa de Endocrinologia, Diabetes e Metabolismo para apresentação deste caso clínico no Congresso Americano de Endocrinologia em 2013 (ENDO 2013: The Endocrine Society's 95th Annual Meeting & Expo).
