






La literatura sobre los inductores en la epilepsia y el trastorno bipolar está contaminada por falsos negativos. Esta segunda parte de una revisión exhaustiva sobre los fármacos antiepilépticos (FAE) con propiedades inductoras aporta más material educativo a los clínicos acerca de la complejidad de interpretar sus interacciones farmacológicas.
Se revisa la farmacología básica de la inducción incluyendo los citocromos P450 (CYP), las enzimas de glucuronización (UGT) y la glucoproteína P (P-gp). Los CYP2B6 y CYP3A4 son muy sensibles a la inducción. El CYP1A2 es moderadamente sensible. Los el CYP2C9 y el CYP2C19 son solo levemente sensibles. El CYP2D6 no puede ser inducida por los fármacos. La inducción de las enzimas metabólicas, los CYP o las UGT, y los transportadores como la P-gp, se debe a un incremento de la síntesis de estas proteínas mediado por los denominados receptores nucleares (receptores constitutivo de androstano, de los estrógenos, de los glucocorticoides y de pregnanoX). Aunque la primera parte de este artículo describe los factores de corrección para los antiepilépticos inductores, la extrapolación de estos valores desde un paciente promedio a un individuo concreto está influenciada por la ruta de administración, la carencia de la enzima metabólica debida a razones genéticas, y la presencia de inhibidores, u otros inductores. También pueden ser importantes las interacciones farmacológicas de los FAE al nivel de los mecanismos farmacodinámicos. Se describen 6 pacientes con una sensibilidad extrema a los inductores antiepilépticos.
The literature on inducers in epilepsy and bipolar disorder is seriously contaminated by false negative findings. Part II of this comprehensive review on antiepileptic drug (AED) inducers provides clinicians with further educational material about the complexity of interpreting AED drug-drug interactions.
The basic pharmacology of induction is reviewed including the cytochrome P450 (CYP) isoenzymes, the Uridine Diphosphate Glucuronosyltransferases (UGTs), and P-glycoprotein (P-gp). CYP2B6 and CYP3A4 are very sensitive to induction. CYP1A2 is moderately sensitive while CYP2C9 and CYP2C19 are only mildly sensitive. CYP2D6 cannot be induced by medications. Induction of UGT and P-gp are poorly understood. The induction of metabolic enzymes such as CYPs and UGTs, and transporters such as P-gp, implies that the amount of these proteins increases when they are induced; this is almost always explained by increasing synthesis mediated by the so-called nuclear receptors (constitutive androstane, estrogen, glucocorticoid receptors and pregnaneX receptors). Although parti provides correction factors for AEDs, extrapolation from an average to an individual patient may be influenced by administration route, absence of metabolic enzyme for genetic reasons, and presence of inhibitors or other inducers. AED pharmacodynamic DDIs may also be important. Six patients with extreme sensitivity to AED inductive effects are described.
La literatura neuropsicofarmacológica sobre las interacciones farmacológicas con los inductores metabólicos de los fármacos está seriamente contaminada con hallazgos falsos negativos. Se niegan sistemáticamente, o al menos se minusvaloran, los efectos de los inductores y la literatura publicada sobre fármacos antiepilépticos (FAE)1 y sobre el trastorno bipolar2 no recalca su relevancia clínica. Esta segunda parte del artículo aporta información para que los clínicos puedan compensar los errores sobre los inductores de los FAE, que aparecen en la literatura. La partei trataba el tema de los inductores potentes (tabla 1) y leves (tabla 2), aportando recomendaciones clínicas sobre la corrección del efecto de los inductores mediante la modificación de la dosis de los estratos inducidos, utilizando factores de corrección (tabla 3)1. Como la literatura de la que se dispone para el cálculo de dichos factores de corrección es altamente limitada, el autor reconoce la probabilidad de que en un plazo de 5años este artículo de revisión pueda estar obsoleto, debiendo modificarse ampliamente los factores de corrección.
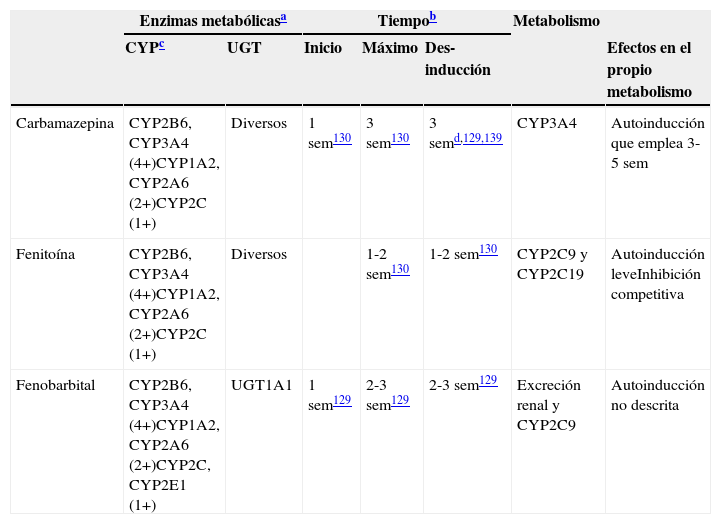
Características de los inductores potentes
| Enzimas metabólicasa | Tiempob | Metabolismo | |||||
|---|---|---|---|---|---|---|---|
| CYPc | UGT | Inicio | Máximo | Des-inducción | Efectos en el propio metabolismo | ||
| Carbamazepina | CYP2B6, CYP3A4 (4+)CYP1A2, CYP2A6 (2+)CYP2C (1+) | Diversos | 1 sem130 | 3 sem130 | 3 semd,129,139 | CYP3A4 | Autoinducción que emplea 3-5 sem |
| Fenitoína | CYP2B6, CYP3A4 (4+)CYP1A2, CYP2A6 (2+)CYP2C (1+) | Diversos | 1-2 sem130 | 1-2 sem130 | CYP2C9 y CYP2C19 | Autoinducción leveInhibición competitiva | |
| Fenobarbital | CYP2B6, CYP3A4 (4+)CYP1A2, CYP2A6 (2+)CYP2C, CYP2E1 (1+) | UGT1A1 | 1 sem129 | 2-3 sem129 | 2-3 sem129 | Excreción renal y CYP2C9 | Autoinducción no descrita |
4+: inducción masiva; 2+: inducción moderada; 1+: inducción leve; sem: semanas.
Constituyen tiempos aproximados incluidos en los artículos de revisión129,130. Los lectores deben ser conscientes de que se han realizado pocos estudios para verificar estos tiempos.
No todos los CYP tienen la misma capacidad de ser inducidos por los inductores potentes. Se incluyen más detalles en la tabla 5. Se piensa que los inductores potentes tienen efectos masivos (4+) sobre CYP2B6 y CYP3A4. Por otro lado, los inductores potentes tienen efectos únicamente leves o moderados sobre la subfamilia CYP2C, que incluye a CYP2C8, CYP2C9 y CYP2C19131. Aunque la literatura no es específica en este punto, el autor piensa que CYP1A2 puede ser inducido de modo intermedio entre los efectos potentes sobre CYP2B6 y CYP3A4 y los efectos leves sobre la subfamilia CYP2C, describiéndose como moderado (2+). Existe información limitada sobre CYP2A6, lo que sugiere un potencial de inducción moderada (2+), aunque los clínicos deben ser conscientes de que pocos fármacos son metabolizados por CYP2A6; esta es la ruta metabólica principal de la nicotina. Existe poca información acerca de CYP2E1, que puede tener un potencial leve para la inducción, aunque los clínicos deben ser conscientes de que pocos fármacos son metabolizados por CYP2E1, aunque es una ruta metabólica menor del alcohol y ciertos fármacos antiepilépticos.
La pérdida de inducción puede emplear más tiempo en los sustratos de CYP1A2 que en los de CYP3A (las vidas medias de inducción respectivas fueron de 105 y 70h, o de 4,4 y 2,9días132.
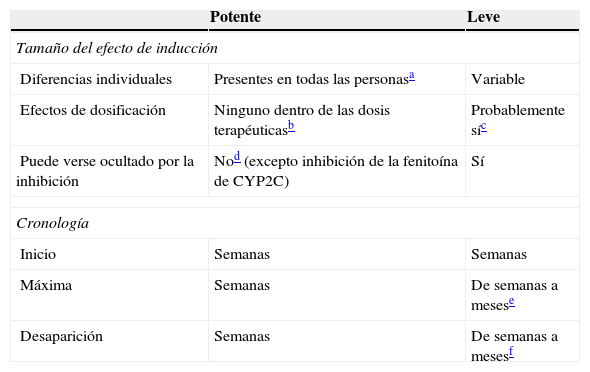
Inductores leves: comparación con los inductores potentes
| Potente | Leve | |
|---|---|---|
| Tamaño del efecto de inducción | ||
| Diferencias individuales | Presentes en todas las personasa | Variable |
| Efectos de dosificación | Ninguno dentro de las dosis terapéuticasb | Probablemente síc |
| Puede verse ocultado por la inhibición | Nod (excepto inhibición de la fenitoína de CYP2C) | Sí |
| Cronología | ||
| Inicio | Semanas | Semanas |
| Máxima | Semanas | De semanas a mesese |
| Desaparición | Semanas | De semanas a mesesf |
Aunque no ha sido estudiado sistemáticamente, se acepta generalmente que los inductores potentes tienden a inducir máximamente a todos los pacientes, siempre que se les hayan administrado dosis mayores que aquellas que causan la inducción máxima.
Se piensa también que una dosis terapéutica para la epilepsia debería causar una inducción máxima en todos los pacientes. Por tanto, el incremento adicional de las dosis superior a las dosis terapéuticas puede no causar más inducción. De igual modo, la administración de otro inductor potente a una persona que toma dosis normales de un inductor potente puede no suponer diferencia alguna.
Consúltese en el texto la información sobre los efectos de la dosificación de la oxcarbazepina, el topiramato y el AVP.
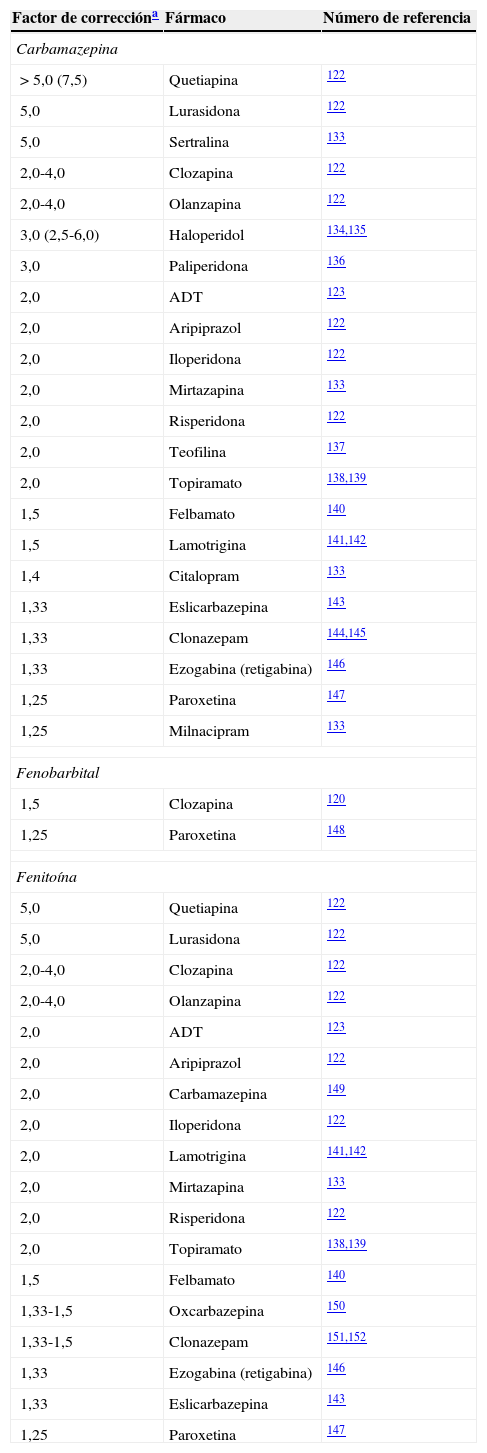
Factores de corrección para inductores potentes
| Factor de correccióna | Fármaco | Número de referencia |
|---|---|---|
| Carbamazepina | ||
| > 5,0 (7,5) | Quetiapina | 122 |
| 5,0 | Lurasidona | 122 |
| 5,0 | Sertralina | 133 |
| 2,0-4,0 | Clozapina | 122 |
| 2,0-4,0 | Olanzapina | 122 |
| 3,0 (2,5-6,0) | Haloperidol | 134,135 |
| 3,0 | Paliperidona | 136 |
| 2,0 | ADT | 123 |
| 2,0 | Aripiprazol | 122 |
| 2,0 | Iloperidona | 122 |
| 2,0 | Mirtazapina | 133 |
| 2,0 | Risperidona | 122 |
| 2,0 | Teofilina | 137 |
| 2,0 | Topiramato | 138,139 |
| 1,5 | Felbamato | 140 |
| 1,5 | Lamotrigina | 141,142 |
| 1,4 | Citalopram | 133 |
| 1,33 | Eslicarbazepina | 143 |
| 1,33 | Clonazepam | 144,145 |
| 1,33 | Ezogabina (retigabina) | 146 |
| 1,25 | Paroxetina | 147 |
| 1,25 | Milnacipram | 133 |
| Fenobarbital | ||
| 1,5 | Clozapina | 120 |
| 1,25 | Paroxetina | 148 |
| Fenitoína | ||
| 5,0 | Quetiapina | 122 |
| 5,0 | Lurasidona | 122 |
| 2,0-4,0 | Clozapina | 122 |
| 2,0-4,0 | Olanzapina | 122 |
| 2,0 | ADT | 123 |
| 2,0 | Aripiprazol | 122 |
| 2,0 | Carbamazepina | 149 |
| 2,0 | Iloperidona | 122 |
| 2,0 | Lamotrigina | 141,142 |
| 2,0 | Mirtazapina | 133 |
| 2,0 | Risperidona | 122 |
| 2,0 | Topiramato | 138,139 |
| 1,5 | Felbamato | 140 |
| 1,33-1,5 | Oxcarbazepina | 150 |
| 1,33-1,5 | Clonazepam | 151,152 |
| 1,33 | Ezogabina (retigabina) | 146 |
| 1,33 | Eslicarbazepina | 143 |
| 1,25 | Paroxetina | 147 |
El factor de corrección del bupropión para la carbamazepina fue de 10,0, calculado por el autor a partir de la limitada información disponible133.
Lamentablemente, pensar que los factores de corrección pueden resolver completamente los problemas de los clínicos en su intento de tratar las cuestiones complejas de la neuropsicofarmacología resultaría una simplificación. Muchas de las combinaciones farmacológicas con las que se encuentran los neurólogos y/o psiquiatras en la práctica clínica diaria no están incluidas en la tabla práctica de la partei, que refleja los factores de corrección de la dosificación (tabla 3). La partei incluye un intento conservador de reflejar la complejidad de la cuestión, mediante la descripción de inductores leves que pueden comportarse también como inhibidores (tabla 4). La parteii constituye un esfuerzo para educar adicionalmente a los clínicos acerca de la naturaleza compleja de la interpretación de las interacciones farmacológicas de los FAE mediante la aportación del conocimiento farmacológico básico para ayudarles a interpretar las interacciones farmacológicas complejas.
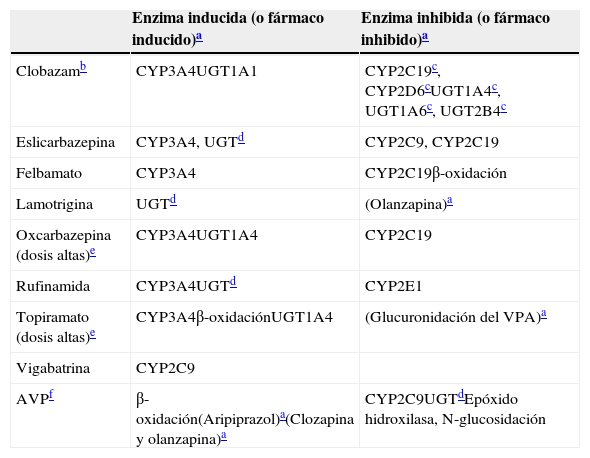
Inductores leves: propiedades inductivas e inhibitorias
| Enzima inducida (o fármaco inducido)a | Enzima inhibida (o fármaco inhibido)a | |
|---|---|---|
| Clobazamb | CYP3A4UGT1A1 | CYP2C19c, CYP2D6cUGT1A4c, UGT1A6c, UGT2B4c |
| Eslicarbazepina | CYP3A4, UGTd | CYP2C9, CYP2C19 |
| Felbamato | CYP3A4 | CYP2C19β-oxidación |
| Lamotrigina | UGTd | (Olanzapina)a |
| Oxcarbazepina (dosis altas)e | CYP3A4UGT1A4 | CYP2C19 |
| Rufinamida | CYP3A4UGTd | CYP2E1 |
| Topiramato (dosis altas)e | CYP3A4β-oxidaciónUGT1A4 | (Glucuronidación del VPA)a |
| Vigabatrina | CYP2C9 | |
| AVPf | β-oxidación(Aripiprazol)a(Clozapina y olanzapina)a | CYP2C9UGTdEpóxido hidroxilasa, N-glucosidación |
AVP: ácido valproico.
Para los fármacos entre paréntesis no se ha establecido con carácter definitivo la enzima tras la inducción o la inhibición.
Aunque esta segunda parte contiene mucha más información teórica que la primera, el autor ha seleccionado la información de acuerdo a su experiencia con las deficiencias de la literatura, para enseñar a los clínicos el modo de navegar sobre las aguas turbulentas de las interacciones farmacológicas con los inductores en la neuropsicofarmacología. Utilizando este enfoque práctico, el autor presenta 7 secciones y aborda 3 cuestiones: farmacología básica, la verdadera complejidad farmacológica de las interacciones entre los fármacos, y la existencia de personas que son inusualmente sensibles a la inducción.
La farmacología básica de la inducción refleja el incremento de producción de las proteínas implicadas en los mecanismos farmacocinéticos. Los inductores incrementan el metabolismo de muchos de los fármacos neuropsicofarmacológicos metabolizados por los isoenzimas del citocromo P450 (CYP), que son las más importantes enzimas oxidativas. Los inductores incrementan a veces la actividad de otras enzimas metabólicas menos comprendidas, las uridina difosfato glucuronosiltransferasas (UGT), que constituyen las enzimas de conjugación más importantes. Recientemente ha sido aclarado que, además de las enzimas metabólicas, otro gran grupo de proteínas farmacocinéticas denominadas transportadores, que normalmente actúan en conjunción con las enzimas metabólicas para eliminar los xenobióticos del organismo, también pueden ser inducidas. Se describe brevemente la glucoproteína P (P-gp) que es el transportador más importante. El área de la literatura que describe a los receptores nucleares está creciendo rápidamente y está comenzando a aportar algo nuestro conocimiento sobre cómo los inductores incrementan la actividad de los CYP, las UGT y la P-gp.
Para resumir, la farmacología básica representa las primeras 4 secciones: a)CYP; b)UGT; c)P-gp, y d)receptores nucleares. Las secciones quinta y sexta abordan la segunda cuestión: la complejidad de la interpretación de la inducción en pacientes sometidos a un régimen de polifarmacia (que constituye la norma en pacientes con epilepsia y trastorno bipolar). Describen, respectivamente, que deben comprenderse los efectos inductivos en el contexto de la complejidad de la farmacocinética en situaciones de polifarmacia y de las interacciones farmacológicas de tipo farmacodinámico. La séptima sección no proviene de la literatura, sino de la práctica clínica del autor. Reconoce que, en raras ocasiones, los clínicos se encuentran con pacientes que son muy sensibles a la inducción y que pueden precisar incrementos masivos de algunos fármacos.
CYPLa terminología de los CYP puede resultar confusa para los clínicos, ya que la nomenclatura de los mismos incluye un número para la familia, una letra para la subfamilia y otro número para el isoenzima específico. Las primeras 3 familias de los CYP se localizan principalmente en el hígado, y están implicadas en el metabolismo de los xenobióticos, por ejemplo los fármacos3. También son importantes para la activación y desactivación de los carcinógenos, y pueden tener cierta participación no bien entendida en el metabolismo endógeno3. Las familias de los CYP superiores a «3» parecen estar principalmente implicadas en el metabolismo endógeno de moléculas complejas incluyendo el colesterol y sus derivados, los esteroides3.
Las 3 primeras familias de los CYP forman parte de las enzimas oxidativas (tradicionalmente denominadas enzimas metabólicas de fasei). El CYP34A4 es el CYP hepático más importante, y representa más de un tercio de los CYP hepáticos. Otros 5 CYP hepáticos, el CYP1A2, el CYP2B6, el CYP2C9, el CYP2C19 y el CYP2D6, son definitivamente importantes para el metabolismo de los fármacos neuropsicofarmacológicos (tabla 5), y es posible que los clínicos que no estén familiarizados con ellos deban realizar un gran esfuerzo para aprender sus nombres y separar uno de otro, dado su nomenclatura confusa.
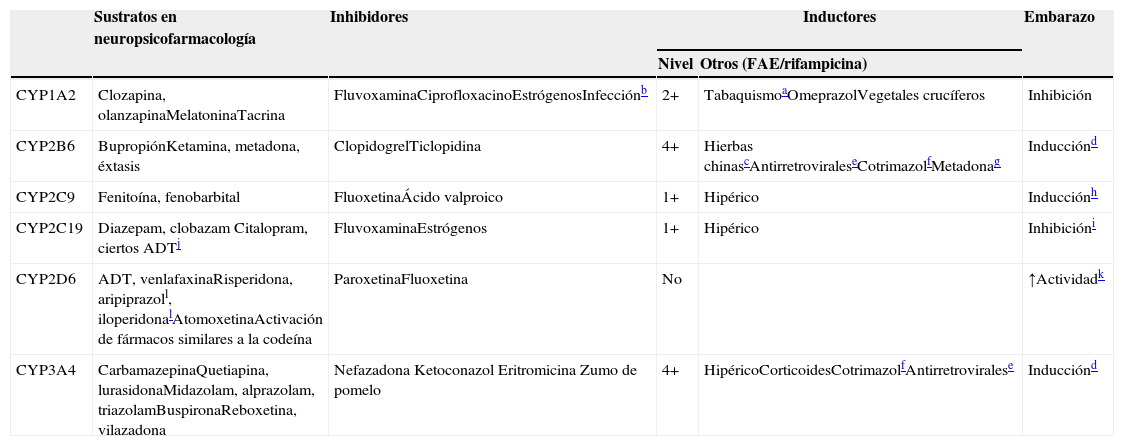
CYP implicadas en el metabolismo de los fármacos
| Sustratos en neuropsicofarmacología | Inhibidores | Inductores | Embarazo | ||
|---|---|---|---|---|---|
| Nivel | Otros (FAE/rifampicina) | ||||
| CYP1A2 | Clozapina, olanzapinaMelatoninaTacrina | FluvoxaminaCiprofloxacinoEstrógenosInfecciónb | 2+ | TabaquismoaOmeprazolVegetales crucíferos | Inhibición |
| CYP2B6 | BupropiónKetamina, metadona, éxtasis | ClopidogrelTiclopidina | 4+ | Hierbas chinascAntirretroviraleseCotrimazolfMetadonag | Inducciónd |
| CYP2C9 | Fenitoína, fenobarbital | FluoxetinaÁcido valproico | 1+ | Hipérico | Inducciónh |
| CYP2C19 | Diazepam, clobazam Citalopram, ciertos ADTj | FluvoxaminaEstrógenos | 1+ | Hipérico | Inhibicióni |
| CYP2D6 | ADT, venlafaxinaRisperidona, aripiprazoll, iloperidonalAtomoxetinaActivación de fármacos similares a la codeína | ParoxetinaFluoxetina | No | ↑Actividadk | |
| CYP3A4 | CarbamazepinaQuetiapina, lurasidonaMidazolam, alprazolam, triazolamBuspironaReboxetina, vilazadona | Nefazadona Ketoconazol Eritromicina Zumo de pomelo | 4+ | HipéricoCorticoidesCotrimazolfAntirretroviralese | Inducciónd |
4+: inducción masiva; 2+: inducción moderada; 1+: inducción leve; FAE: fármaco antiepiléptico.
Los hidrocarburos aromáticos policíclicos del humo tienen efectos inductivos. Estos compuestos se encuentran también en los alimentos a la parrilla y los granos del café tostado, que pueden tener también efectos inductivos22.
Las infecciones respiratorias, otras infecciones graves, tales como la pielonefritis o apendicitis, o incluso las inflamaciones graves, pueden inhibir CYP1A2, porque las citoquinas liberadas inhiben CYP1A2.
El ferulato de sodio se comportó como un inductor del metabolismo del bupropión en un estudio. Se trata de la sal sódica del ácido ferúlico, que está ampliamente distribuida en hierbas y fórmulas chinas como Ligusticum, Chuanxiong y Chaihu-Sugan-San14. Otro inductor es la baicalina, un glucurónico flavonoide extraído de la planta médica Radix scutellariae, que está presente en frutas, vegetales y bebidas derivadas de las plantas (té, vino tinto), y gran variedad de hierbas medicinales, que incluyen: Huang-Lian-Jie-Du-Tang, hangeshashinto, San-Huang-Xie-Xin-Tang, Da-Chai-Hu-Tang y Xiao-Chai-Hu-Tang15.
El embarazo induce definitivamente a CYP2B6 y CYP3A425. Conforme a un estudio in vitro, CYP2B6 y CYP3A4 son inducidas tanto por los estrógenos como por la progesterona. La progesterona induce también a CYP3A5.
Los estudios in vitro indican que la metadona puede inducir su propio metabolismo, con la mediación no solo de CYP2B6 sino también de CYP3A420.
Se cree que CYP2C9 se incrementa durante el embarazo debido al incremento de la eliminación de fenitoína. No puede descartarse que mecanismos diferentes a la inducción de CYP2C9 puedan explicar los cambios en la eliminación de la fenitoína durante el embarazo. El estradiol incrementa la actividad de CYP2C9 sin afectar a la expresión, mediante mecanismos desconocidos26.
Se cree que los estrógenos son inhibidores competitivos de CYP2C19, aunque un estudio reciente sugería que pueden inhibir la expresión de CYP2C1912.
CYP2C19 es la enzima principal para la demetilación de la amitriptilina, la clomipramina y la imipramina. A continuación sus metabolitos son metabolizados mediante hidroxilación, principalmente por CYP2D6.
No se comprende bien la causa del posible incremento de la actividad de CYP2D6 durante el embarazo, ya que se cree que CYP2D6 no puede ser inducida. Un estudio reciente sugería que el embarazo puede eliminar un supresor de la expresión de CYP2D624.
Existen múltiples isoenzimas CYP extrahepáticas implicados en el metabolismo xenobiótico y no incluidas en la tabla 5, aunque son menos conocidos y menos importantes. Los 2 CYP extrahepáticos más importantes pueden ser el CYP3A4 y el CYP3A5. El CYP3A4 es también el CYP más importante a nivel del intestino delgado4. El CYP3A4 del intestino delgado (con la P-gp; véase dicha sección) forma parte de lo que los farmacólogos denominan el metabolismo de primer paso. Dicho metabolismo de primer paso5 hace referencia a la disminución sustancial de ciertos fármacos al utilizar la vía de administración oral en lugar de la intravenosa (i.v.). El metabolismo a nivel del intestino y la primera vez que atraviesa el hígado es lo que se denomina metabolismo de primer paso. Aunque algunos fármacos que exhiben un metabolismo de primer paso son metabolizados por las UGT (p.ej., la asenapina), existe un consenso en cuanto a que el CYP3A4 (y la P-gp) localizadas en el intestino e hígado explican el metabolismo de primer paso para los sustratos del CYP3A4. El CYP3A5 es homóloga al CYP3A4, parece metabolizar muchos de los mismos sustratos que CYP3A4 y es particularmente importante para el metabolismo renal de los fármacos. En general, para la mayoría de los fármacos metabolizados por la subfamilia CYP3A, se cree que la contribución relativa del CYP3A5 a su metabolismo total es pequeña6.
La columna de sustratos de la tabla 5 describe qué fármacos en neuropsicofarmacología7,8 son metabolizados por los 5 CYP hepáticos más importantes. Otra columna describe los inhibidores de los CYP9–13. Los inhibidores actúan normalmente fijándose al CYP e inactivando esa molécula. La tabla se centra en los inhibidores potentes que no pueden ser desplazados fácilmente por los sustratos, aunque cualquier sustrato en las circunstancias clínicas adecuadas puede comportarse como un inhibidor competitivo clínicamente relevante de sus enzimas metabolizadoras. Ciertos inhibidores de los CYP, en particular las hormonas sexuales femeninas, pueden no solo inactivar algunos CYP (el CYP1A2 y el CYP2C19), sino también hacer disminuir su síntesis12.
La tercera columna de la tabla 5 describe el nivel de inducción, que hace referencia a cuán sensible es el CYP a la inducción (véase la sección sobre la farmacocinética compleja de la polifarmacia). La siguiente columna incluye otros inductores9,14–22aparte de los FAE (véanse los CYP inducidas por los FAE en las tablas 1 y 2). No se incluye la rifampicina para los 5 CYP, ya que es un inductor promiscuo de dichos CYP (y de otras enzimas). La última columna se centra en los efectos complejos del embarazo, que pueden incrementar o disminuir la actividad de dichos CYP12,23–26.
Variaciones genéticasLas 3 primeras familias de los CYP incluyen genes que son altamente polimórficos, lo que significa que son frecuentes las variaciones genéticas que afectan a su función. Aunque no se comprenden bien, las variaciones genéticas de las primeras familias de los CYP, pueden estar relacionadas con las presiones de la evolución en diferentes ambientes con la exposición a diferentes xenobióticos, particularmente en la dieta. Es probable que nuestros ancestros, en tiempos de carestía, no tuvieran más elección que la de comer cualquier planta que pudieran encontrar para paliar su hambre. Muchas plantas incluyen toxinas para protegerse de los animales herbívoros. Como muchos fármacos se derivan de las plantas, no es sorprendente que utilicemos los mismos CYP para metabolizar los fármacos. Los clínicos deben saber que las primeras 3 familias de los CYP varían de una especie a otra; por ello, los estudios animales de los CYP no pueden extrapolar a los humanos. Por lo que se utilizan los estudios in vitro con hepatocitos humanos para estudiar estas 3 primeras familias en el laboratorio.
Las empresas bombardean a menudo a los clínicos remarcando los beneficios de la genotipificación de los CYP. El autor piensa que para interpretar los resultados de la genotipificación los médicos clínicos precisan un conocimiento sofisticado de los alelos de los CYP, así como de las limitaciones técnicas de las diversas técnicas sobre genotipificación27–29.
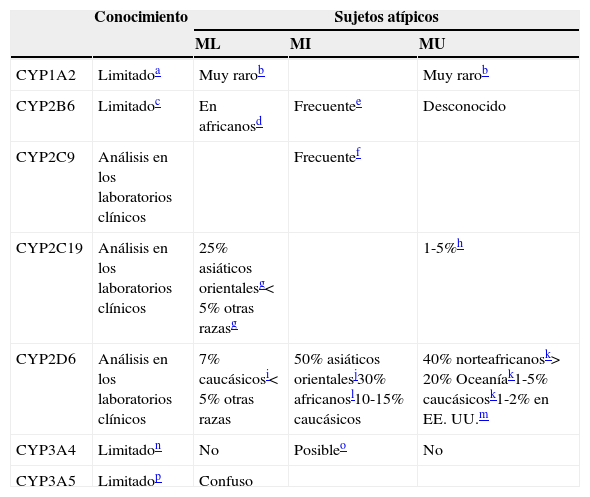
Como se resume en la tabla 6, la opinión del autor es que muchas variaciones de los CYP no están preparadas para el uso clínico29. Solo 3 de ellas, las del CYP2C9, las del CYP2C19 y las del CYP2D6, pueden estar preparadas, aunque las del CYP2C9 tiene aplicaciones relativamente pequeñas en la neuropsicofarmacología. La genotipificación de los CYP1A230,31, CYP2B632,33, CYP3A434 y CYP3A534,35se describe como limitada, ya que nuestra comprensión acerca de sus variaciones genéticas y de las relaciones genotipo-fenotipo es escasa. Es fácil encontrar artículos que remarquen el potencial de la genotipificación de los CYP, resaltando los estudios con resultados significativos, e ignorando los estudios que no los tengan y las grandes lagunas en la comprensión de las relaciones genotipo-fenotipo. En el otro extremo, podemos encontrarnos con investigadores con un enfoque basado en la evidencia que «socavan» la genotipificación de los CYP explicando que su función es muy pequeña en la prescripción de los inhibidores selectivos de la recaptación de serotonina (ISRS). El pequeño papel que juega la genotipificación de los CYP en la dosificación de muchos ISRS tiene poca relevancia en la materia, ya que cualquier farmacólogo que maneje los ISRS podría haberlo predicho utilizando su conocimiento sobre los mecanismos farmacológicos, en lugar de realizar una revisión sistemática de la literatura36. Independientemente de ello, se ha recomendado la realización de correcciones de la dosificación, con arreglo a la genotipificación del CYP2C19 para citalopram, escitalopram y sertralina29.
Implicación de CYP en el metabolismo de los fármacos: variaciones genéticas
| Conocimiento | Sujetos atípicos | |||
|---|---|---|---|---|
| ML | MI | MU | ||
| CYP1A2 | Limitadoa | Muy rarob | Muy rarob | |
| CYP2B6 | Limitadoc | En africanosd | Frecuentee | Desconocido |
| CYP2C9 | Análisis en los laboratorios clínicos | Frecuentef | ||
| CYP2C19 | Análisis en los laboratorios clínicos | 25% asiáticos orientalesg<5% otras razasg | 1-5%h | |
| CYP2D6 | Análisis en los laboratorios clínicos | 7% caucásicosi<5% otras razas | 50% asiáticos orientalesj30% africanosl10-15% caucásicos | 40% norteafricanosk>20% Oceaníak1-5% caucásicosk1-2% en EE.UU.m |
| CYP3A4 | Limitadon | No | Posibleo | No |
| CYP3A5 | Limitadop | Confuso | ||
MI: metabolizador intermedio; ML: metabolizador lento; MU: metabolizador ultrarrápido.
Ciertos estudios indican que algunos alelos pueden influir en la inducción. CYP1A2*1C ha sido asociada a una baja inducibilidad y CYP1A2*1F a una elevada inducibilidad, aunque no existe un acuerdo generalizado en la literatura30,31.
Los artículos de revisión del CYP1A231 reportan normalmente que no existen ML claramente identificados por no tener la enzima, ni MU con un incremento claro de la actividad. Raramente, la literatura sobre clozapina describe a algunos sujetos extremos compatibles con ser ML del CYP1A2 (p.ej., un paciente con un alelo CYP1A2*7)41. Se han descrito ciertos casos con un perfil de MU, aunque no se han identificado unas variaciones genéticas claras42,43.
La actividad del CYP2B6 varía de acuerdo con un gran número de variaciones genéticas del CYP2B6, que incluyen los polimorfismos de un único nucleótido, y haplotipos33.
CYP2B6*18 se produce predominantemente en africanos y no expresa una proteína funcional, al menos para algunos sustratos. Las frecuencias de personas con deficiencia de un alelo se describen como del 5-12% en africanos y del 4-8% en afroamericanos14. Por tanto, aunque no se describen las frecuencias de ML en los artículos, se pueden predecir dichas frecuencias <1% en estas poblaciones.
CYP2B6*6 es probablemente el alelo con bajo funcionamiento y mayor frecuencia, produciéndose en cerca del 15 al 60% de las diferentes poblaciones32.
Algunos alelos frecuentes del CYP2C9 tienen actividad reducida. CYP2C9*1 es el alelo normal, y un sujeto con *1/*1 tendrá una actividad de 1,0. CYP2C9*2 tiene menor actividad, según Castellan et al.40; un sujeto con *2/*2 tendrá una actividad de 0,70, mientras que un sujeto con *1/*2 tendrá una actividad de 0,82. CYP2C9*3 tiene muy baja actividad; según Castellan et al.40, un sujeto con *3/*3 tendrá una actividad de 0,13, mientras que un sujeto con *1/*3 tendrá una actividad de 0,56, y *2/*3 tendrá una actividad de 0,39. Por tanto, para *3/*3 se recomienda una reducción de la dosis de fenitoína del 65%, y del 85% de fenobarbital.
Para el CYP2C19, los alelos inactivos más importantes son *2 y *3, que son frecuentes en asiáticos orientales38.
CYP2C19*17 se asocia a un incremento de la actividad. Scott et al. (2011)52 describieron que el 21% de los europeos y el 18% de los africanos tenían un alelo CYP2C19*17. Con estas frecuencias puede estimarse que aproximadamente el 4% de los europeos y el 3% de los africanos tendrían 2 alelos con incremento de actividad, pudiéndose considerar MU del CYP2C19 (CYP2C19*17/CYP2C19*17). Otros autores argumentan que el CYP2C19*17 puede ser irrelevante para la mayoría de los fármacos53.
Los alelos inactivos más importantes del CYP2D6 son *3, *4, *5 (supresión) y *638. Los diferentes artículos de revisión aportan prevalencias diferentes de los ML de CYP2D6. En una revisión reciente, McGraw y Waller45 describieron un 3-10% en caucásicos, un 0-19% en afroamericanos, un 6,0% en hispanos, un 0-4,4% en americanos nativos y un 0,3% en asiáticos orientales.
En CYP2D6, las personas con una actividad metabólica menor de lo normal se llaman MI, aunque la terminología varía de un laboratorio a otro. A veces, los resultados de los análisis de laboratorio del CYP2D6 describen a los sujetos con al menos un alelo normal como MI, mientras que otros los considerarán metabolizadores normales27,38. El alelo *10 es frecuente en los asiáticos orientales. Suponiendo que 2 alelos activos normales tienen una actividad de 1, un alelo activo y *10 tienen un nivel de actividad de 0,54 y 2 *10 solo 0,1049.
CYP2D6*17 se halla en individuos con antepasados africanos, que normalmente tienen una actividad menor, aunque una actividad normal para la risperidona37.
En un estudio con una muestra grande en un hospital psiquiátrico de Estados Unidos37, la prevalencia de los MU del CYP2D6 fue del 1,5% (intervalo de confianza del 95% [IC95%]: 1,1-1,9). En afroamericanos fue del 2,0% (IC95%: 1,1-3,7) para muchos fármacos, pero como el CYP2D6*17 puede tener una actividad normal para la risperidona, la frecuencia de los MU para la risperidona fue ligeramente superior, es decir, del 2,9% (IC95%: 1,7-4,9).
La expresión y la actividad del CYP3A4 varían ampliamente entre los individuos, aunque se cree que se debe no solo a los factores genéticos sino a los no genéticos, tales como los hormonales y el estado de salud, y el impacto de los estímulos ambientales. Muchas de las variaciones genéticas descritas no tienen consecuencias funcionales, o estas son muy raras para poder contribuir de algún modo a la variabilidad de CYP3A434.
La literatura sobre el CYP3A5 es bastante confusa. La historia tradicional es que existen 3 alelos con nula o baja expresión: CYP3A5*3, CYP3A5*6 y CYP3A5*7. CYP3A5*6 y CYP3A5*7 solo se encuentran en personas con antepasados africanos. Las personas se clasifican normalmente como expresores altos o bajos de CYP3A5. Las frecuencias aproximadas de los expresores altos son del 70% en africanos y del 10% en caucásicos34. Más recientemente, Bains et al.35 reportaron que no se ha establecido definitivamente que CYP3A5*6 esté asociada a una expresión menor; incluso suponiendo que el número de expresores altos en la población africana varíe ampliamente, su media puede ser del 43%. Para confundir aún más la historia de CYP3A5, la relevancia de su polimorfismo puede variar de un fármaco a otro, no habiéndose establecido de manera definitiva34.
Por tanto, a pesar de que el autor ha genotipado más de 5.000 pacientes por el CYP2D6 y/o el CYP2C1937, él piensa que la genotipificación de los CYP debe utilizarse juiciosamente en personas específicas con respuestas inusuales, que podrían ser compatibles con anomalías genéticas del CYP2D6 y/o el CYP2C19. Esta genotipificación debería combinarse con información sobre los inhibidores e inductores. Según la experiencia del autor, la genotipificación del CYP2D6 y del CYP2C19 puede ser ocasionalmente útil en pacientes con una respuesta peculiar a ciertos fármacos psiquiátricos38, en particular los fármacos psiquiátricos de primera generación, ya que los antidepresivos tricíclicos (ATC) y las fenotiazinas son principalmente metabolizados por el CYP2D6; estos fármacos tienen ventanas terapéuticas estrechas. De hecho, se han desarrollado excelentes directrices sobre dosificación de los ATC, basadas en la genotipificación del CYP2D6 y el CYP2C197. Con los antipsicóticos y los antidepresivos de segunda generación, la genotipificación de los CYP es menos útil, ya que poseen rutas metabólicas muy diferentes9,13,39. Se ha recomendado realizar correcciones de dosificación, conforme a la genotipificación del CYP2D6 para venlafaxina, aripiprazol, risperidona, zuclopentixol y atomoxetina29. Pacientes y colegas consultan a menudo al autor acerca de algunos sujetos extremos que no toleran o no responden a muchos fármacos de la misma clase. A menudo, dichas situaciones no se pueden explicar por medio de las variaciones genéticas de los CYP, sino por otros factores38.
Las variaciones genéticas más importantes dentro de las 3 primeras familias de los CYP se describen en la tabla 627,31,32,37,38,40–54. La terminología de la genotipificación y fenotipificación de los CYP es bastante confusa para los clínicos. Su complejidad se debe al complejo desarrollo histórico de la nomenclatura de los CYP55. En primer lugar, se describió el concepto de metabolizadores lentos (ML). Quedó claro que para el CYP2D6 y el CYP2C19 existían algunos sujetos que parecían tener una actividad metabólica baja. Una de las primeras descripciones hizo referencia a los ATC; algunos sujetos tenían muy poca capacidad para metabolizar los ATC55. Posteriormente se descubrió que los ATC eran metabolizados por el CYP2D6; los ML del CYP2D6 eran individuos que no tenían CYP2D6 en el hígado. Los ML tienen 2 alelos inactivos del CYP2D6 (un alelo inactivo en el cromosoma del padre y otro alelo inactivo en el otro cromosoma, el de la madre), de modo que no se produce proteína del CYP2D6 o, de producirla, es completamente inactiva. Así, los fenotipos del CYP2D6 incluyeron inicialmente a los ML, que no tienen CYP2D6 en su cuerpo, y a las personas normales a las que se denominaron metabolizadores rápidos (MR). El fenotipo del CYP2D6 en la población se describió inicialmente como bimodal, un modo para los ML y otro para los MR. Posteriormente se descubrió que los metabolizadores ultrarrápidos (MU) en una familia en Suecia que tenían niveles indetectables de los ATC a pesar de tomar dichos fármacos y que tenían más CYP2D6 de lo normal56. Tenían de 3 a 13 alelos activos del CYP2D6. Un paciente con 3 alelos recibió un alelo normal de uno de los padres y 2 alelos activos del otro (el alelo se había duplicado). Un paciente con 13 alelos recibió un alelo normal de uno de los padres y 12 copias activas del otro (el alelo se había multiplicado por 12).
También se ha descubierto que el CYP2C19 es polimórfico incluyendo ML y MR55, aunque tiene una distribución racial diferente al CYP2D6 (tabla 6). Posteriormente se describieron también los MU del CYP2C19; que tenían un alelo que está asociado a una expresión mayor del CYP2C19. Por tanto, los mecanismos genéticos que explican los MU pueden incluir la multiplicación de un gen activo (que se incluye normalmente en el concepto de variación del número de copias) como el CYP2D6, o un alelo con un incremento de expresión, como en el CYP2C19.
Los clínicos deben recordar que una persona con un genotipo MR puede tener un fenotipo de ML cuando toma un inhibidor potente que elimina toda la actividad de la enzima. Una persona puede tener un fenotipo de MU cuando toma un inductor potente que incrementa la actividad de la enzima relevante.
En el CYP2C9 no se hallaron ML, sino sujetos con baja actividad. En algunas personas, en particular los asiáticos orientales, se ha detectado en el CYP2D6 individuos con baja actividad. Se les denominó metabolizadores intermedios (MI). Por tanto, para el CYP2D6 y el CYP2C19 las variaciones genéticas difieren de una raza a otra, e incluyen sujetos que van desde los que no tienen actividad (ML) a los que tienen poca actividad (MI), a los normales (MR) y hasta los que tienen excesiva actividad (MU) (tabla 6).
Algunos sujetos raros, no bien estudiados en la literatura, son relevantes para los prescriptores de neuropsicofarmacología; son doblemente ML para el CYP2D6 y el CYP2C1957. Esta variante genética combinada es muy rara (<1/1.000), aunque puede ser muy relevante para la prescripción de antidepresivos. Muchos antidepresivos orales son metabolizados por el CYP2D6 y/o el CYP2C19; estos raros dobles ML, según la experiencia del autor, tienen una respuesta negativa con muchos antidepresivos, ya que no los pueden tolerar bien a dosis normales. Sin embargo, podrían tolerar las dosis normales de mirtazapina, que no depende del CYP2D6 o del CYP2C19 para su metabolismo57, o dosis normales de bupropión, que es metabolizado principalmente por el CYP2B6.
Los clínicos deben ser conscientes de que se está empezando a conocer una nueva área genética, relevante para la función de los CYP, aunque no se ha establecido bien aún su relevancia clínica. La CYP oxidorreductasa (POR) es una enzima microsómica localizada cerca de los CYP; influye en la función de los CYP mediante la donación de electrones. Recientemente se ha propuesto que las variaciones genéticas de la POR pueden resultar mejores predictores del fenotipo del CYP3A4 que las variaciones genéticas del CYP3A458, pudiendo asociarse las variaciones genéticas de la POR a un incremento o disminución de la actividad de los CYP de estas 3 primeras familias59.
UGTLas UGT son las enzimas más importantes de entre las enzimas de conjugación (tradicionalmente denominadas enzimas metabólicas de faseii), siendo las principales enzimas metabólicas para una serie de antipsicóticos, FAE y benzodiazepinas (tabla 7). Las UGT han recibido menos atención y su función es menos comprendida que la de los CYP60. El conocimiento de las UGT se sitúa unos 10-15años por detrás de los CYP, hasta el punto de que la información suministrada en esta revisión sobre los efectos inductivos de las UGT es mucho menos fidedigna que la de los inductores de los CYP, y tienen mucha más probabilidad de precisar correcciones y actualizaciones en los próximos 5-10años. Los factores que contribuyen a dicha ignorancia incluyen: a)la dificultad para desarrollar métodos analíticos de medición de los glucurónidos; b)la superposición de la actividad de las UGT y la falta de sondas selectivas; c)la complejidad del ciclo de glucuronidación, que incluye la reabsorción a través del ciclo enterohepático y la participación en la desconjugación por parte de las β-glucuronidasas, que también están presentes en las bacterias del intestino, y d)los compuestos endógenos que son frecuentemente sustratos, inhibidores o inductores de las UGT; esto se produce con mucha más frecuencia que en los CYP, lo que aporta un nivel adicional de complejidad a las interacciones farmacológicas61,62.
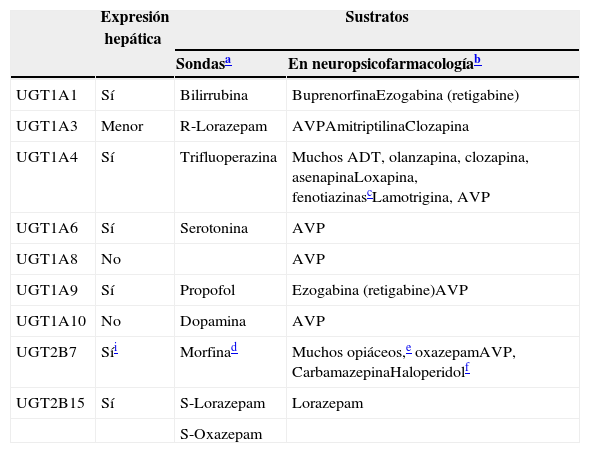
Participación de las UGT en el metabolismo de los fármacos
| Expresión hepática | Sustratos | ||
|---|---|---|---|
| Sondasa | En neuropsicofarmacologíab | ||
| UGT1A1 | Sí | Bilirrubina | BuprenorfinaEzogabina (retigabine) |
| UGT1A3 | Menor | R-Lorazepam | AVPAmitriptilinaClozapina |
| UGT1A4 | Sí | Trifluoperazina | Muchos ADT, olanzapina, clozapina, asenapinaLoxapina, fenotiazinascLamotrigina, AVP |
| UGT1A6 | Sí | Serotonina | AVP |
| UGT1A8 | No | AVP | |
| UGT1A9 | Sí | Propofol | Ezogabina (retigabine)AVP |
| UGT1A10 | No | Dopamina | AVP |
| UGT2B7 | Síi | Morfinad | Muchos opiáceos,e oxazepamAVP, CarbamazepinaHaloperidolf |
| UGT2B15 | Sí | S-Lorazepam | Lorazepam |
| S-Oxazepam | |||
ATC: antidepresivos triciclicos. AVP: acido valproico.
Las sondas son fármacos que se utilizan para estudiar la enzima específica, ya que serán sustratos relativamente específicos, aunque para las UGT los diferentes artículos proporcionan diferentes sondas. Solo se ha utilizado un artículo reciente de un experto en la materia73 para desarrollar esta columna.
La morfina es metabolizada a un 3-glucurónido sin efecto analgésico por UGT2B7, UGT1A8 y UGT1A3, y a un 6-glucurónido que es un analgésico más potente que la morfina, principalmente por UGT2B775.
Aparte de la morfina, muchos opiáceos son glucuronizados por UGT2B7, incluyendo buprenorfina, codeína, naloxona y naltrexona. Buprenorfina y naltrexona pueden ser también glucuronizadas por UGT1A160.
Un estudio in vitro reciente76 describió que el haloperidol es glucuronizado principalmente por UGT2B7, con contribuciones menores de UGT1A4 y UGT1A9. Estas UGT serían responsables del 50-60% de la eliminación del haloperidol, frente al 20-30% por parte de CYP3A4.
Las UGT se localizan en la membrana interna de cara al lado luminal del retículo endoplásmico del tejido hepático y extrahepático (en particular piel, pulmones, intestino delgado y riñón), con acceso directo a los metabolitos resultantes de los CYP y otros enzimas oxidativos63. Más recientemente, algunos autores han propuesto que las interacciones proteicas pueden ser importantes para la comprensión de la función de las UGT. Las UGT pueden actuar mediante la formación de complejos de diversas UGT que actúan al mismo tiempo en los substratos de glucuronización62. Si dicha idea acaba siendo verdad, no tendría sentido tratar de establecer qué UGT específica puede ser relevante para la metabolización de un sustrato específico. Además, existe información inicial acerca de que los CYP y UGT puedan interactuar en la metabolización de los fármacos62, y que la función de las UGT puede ser fundamental para la comprensión de las propiedades fisiológicas de muchos inhibidores de los CYP64.
Las UGT transfieren el grupo glucuronil de la uridina-5’ difosfoglucuronato a muchos compuestos lipofílicos. El glucurónido resultante es más soluble en agua, menos tóxico y más fácilmente excretable que el compuesto madre. Los glucurónidos representan la mayoría del material de desecho que se encuentra en la bilis y la orina. Las UGT han evolucionado para catalizar la glucuronidación de ambos compuestos endógenos (bilirrubina, hormonas tiroideas, hormonas sexuales y serotonina) y xenobióticos (p.ej., los acetaminofenos y la morfina se eliminan fundamentalmente de este modo). La importancia biológica de la glucuronidación está limitada por las limitaciones de su investigación, aunque parece razonable postular que sirve principalmente para ayudar a la eliminación de los sustratos del cuerpo65, aunque en ciertos casos puede producir compuestos más activos o tóxicos65,66.
Los genes humanos de las UGT se clasifican en 4 familias, basadas en la identidad secuencial: la familia UGT1 de los glucuronidatos de bilirrubina y fenoles xenobióticos, la familia UGT2 de los glucuronidatos de los esteroides y ácidos biliares. Se desconoce la función de la familia UGT3A, mientras que la familia de la UGT8A tiene solo un gen humano, que puede estar implicado en la biosíntesis de membranas67. Al igual que en los CYP, cada especie parece tener sus propias UGT con diversos sustratos.
La subfamilia humana de UGT1 se deriva de un único gen, con 13 promotores/primer exon y un conjunto de 2 a 5 exones compartidos67. El ARN mensajero que codifica cada isoforma de la UGT se forma por la fusión de un tipo de exón1 con los 4 exones de los exones 2 a 5. Las mutaciones genéticas en las regiones comunes del 2 al 5 pueden originar cambios en la actividad y/o la expresión de todas las isoformas, mientras que las mutaciones genéticas del único exón1, o región promotora, afectan únicamente a la única isoforma implicada68. Por ello, pueden generarse diversas transcripciones potenciales del locus de UGT1A que contienen terminaciones únicas 5’ y terminaciones idénticas 3′, incluyendo 4 enzimas localizadas en el hígado (UGT1A1, UGT1A3, UGT1A4 y UGT1A9) y 3 enzimas menos conocidas (UGT1A7, UGTA18 y UGT1A10) sin expresión hepática, pero con expresión principal en los epitelios gastrointestinales69. La tabla 760,70–76 incluye una descripción de las UGT que son relevantes en neuropsicofarmacología.
La familia UGT2 tiene subfamilias que son sintetizadas por una serie de genes similares localizados en un casete en el cromosoma 4. Tres isoformas específicas olfatorias se incluyen en la subfamilia UGT2A. La subfamilia UGT2B incluye enzimas metabólicas que son responsables de la glucuronidación de los esteroides y los ácidos biliares. La UGT2B tiene 12 miembros, 5 pseudogenes (genes inactivos) y 7 genes. La tabla 7 describe las 2 mejores conocidas, UGT2B7 y UGT2B15.
Variaciones genéticasLas enzimas UGT pueden ser polimórficas, aunque estas variaciones genéticas no son lo suficientemente conocidas como para desarrollar una tabla similar a la de los CYP. Los polimorfismos de la UGT1A1 son los que mejor se conocen. El síndrome de Gilbert es una variación genética asociada a la reducción de los niveles de UGT1A1, que se manifiesta como hiperbilirrubinemia no conjugada y no hemolítica, sin daño estructural hepático77.
Se han identificado muchas variaciones polimórficas en otras UGT, además de la de UGT1A169,78, aunque en la mayoría de los casos no se ha establecido bien si dichas variaciones son o no funcionales, y a menudo no se han replicado los hallazgos asociados a los cambios en la respuesta farmacológica. También pueden ser importantes para los genes de las UGT otros procesos genómicos clave tales como las variaciones en el número de copias, los factores epigenéticos y los mecanismos de escisión79. Para resumir, no es probable que en los próximos 5años se utilice en la clínica otros polimorfismos genéticos de las UGT, además del de la UGT1A1, que ha sido recomendado para ciertos fármacos anticancerígenos, por lo que los clínicos no precisan estar familiarizados con ello.
InhibidoresPuede utilizarse el ácido valproico (AVP) como ejemplo para describir nuestra limitada comprensión de la fisiología de la glucuronidación y la de su inhibición. El conjugado del acil glucurónido (valproato-glucurónido, o VPAG) parece ser el mayor metabolito del AVP en la orina, y representa el 30-50% de la dosis de AVP80. Cinco UGT hepáticas, la UGT1A3, la UGT1A4, la UGT1A6, la UGT1A9 y la UGT2B7, y dos UGT fundamentalmente intestinales, la UGT1A8 y la UGT1A1080,81, podrían estar implicadas en la glucuronidación del AVP. De acuerdo con un estudio in vitro realizado por Argikar y Remmel80, la UGT2B7 posee la mayor actividad metabólica para el AVP, siendo UGT1A6 la segunda, y a continuación el resto de las UGT, con una actividad similar. Argikar y Remmel80 advirtieron que no se sabe cómo extrapolar la relevancia clínica de las UGT extrahepáticas, tales como la UGT1A8 y la UGT1A10, debido a la falta de información acerca de su expresión. Además de ser metabolizado por las UGT, el AVP ser un inhibidor clínicamente relevante de dichas UGT. Sin embargo, no es fácil establecer de modo definitivo, mediante la revisión de la literatura, qué UGT son inhibidas por el AVP.
En la literatura sobre neuropsicofarmacología existe un consenso definitivo en cuanto a que el AVP inhibe la glucuronidación de 2 fármacos, la lamotrigina74,80,82 y el lorazepam74,83. Estos son metabolizados principalmente por las UGT, pero como no existe acuerdo sobre qué UGT es inhibida por el AVP, no puede explicarse los mecanismos de dicha inhibición. Si se acepta que la UGT1A4 es la enzima más importante para el metabolismo de la lamotrigina, y la UGT2B15 para el lorazepam, entonces el VPA es probablemente un inhibidor clínicamente relevante de UGT1A4 y UGT2B15. Otros artículos describen al AVP como inhibidor de las UGT2B784,85, UGT2B15 y UGT1A984. En resumen, el autor no está absolutamente seguro qué UGT puede ser inhibida por el AVP, y la literatura aporta una visión complicada y a veces contradictoria. La literatura no clarifica si la inhibición del fármaco por parte del AVP en un estudio in vitro es un signo de inhibición de una UGT específica, o una inhibición de múltiples UGT. Además, el autor no tiene claro si un estudio in vitro que utiliza el VPA como inhibidor puede extrapolarse a un caso in vivo en el que no solo está presente el VPA sino también los metabolitos del mismo, contribuyendo probablemente a la inhibición de las UGT, y posiblemente a su inducción, como indica la partei de este artículo de revisión. Resumiendo, tenemos una comprensión muy limitada de la inhibición (e inducción) de las UGT.
P-gpLa relevancia de los transportadores para el metabolismo de los fármacos y en las interacciones farmacológicas ha comenzado a comprenderse recientemente, aunque dicha comprensión se ha expandido rápidamente en los últimos 10años. Aunque algunos autores han sugerido que los transportadores deberían considerarse como la faseiii del metabolismo de los fármacos, esta terminología se sigue raramente86. Funcionalmente, los transportadores se clasifican como aquellos que median en la absorción celular de los fármacos (transportadores de absorción o influjo) y los que median en la exportación de los fármacos, o metabolitos de los fármacos, al exterior de las células (transportadores de eflujo)87.
P-gp es el transportador de fármacos mejor estudiado, pero los psiquiatras o neurólogos que revisen la literatura encontrarán que induce a confusión desde un principio, ya que los artículos utilizan 3 nombres diferentes88. P-gp fue descrita inicialmente como una glucoproteína de superficie que modificaba la permeabilidad de la colchicina en el modelo celular animal. Luego se vio que existía un exceso de expresión en las muestras tumorales que se asociaba a la resistencia a múltiples fármacos; el gen era el denominado proteína resistente a múltiples fármacos1 (MDR1). Su elevada homología con los transportadores bacterianos sugería que la P-gp es un transportador de eflujo. Los estudios inmunohistoquímicos demostraron la expresión de P-gp en tejidos con funciones secretoras o excretoras (hígado, riñón y tracto gastrointestinal) y en localizaciones de las barreras sangre-tejido, tales como la barrera hemato-encefálica (BHE). Este patrón de expresión indicaba que la P-gp puede influir sobre la respuesta y toxicidad xenobiótica. Se vio que la P-gp formaba parte de una familia muy grande de genes transportadores que se fijan al ATP, algunos de ellos implicados en los trastornos genéticos humanos86, y fue renombrada como miembro1 de la subfamiliaB del casete que se fija al ATP (ABCB1). Por tanto, los artículos sobre esta materia utilizan 3 encabezamientos para la misma proteína o gen productor de la proteína: P-gp, MDR1 o ABCB1.
P-gp juega un papel central en la absorción, distribución y excreción de una amplia variedad de fármacos. La Pg-p se expresa en diversos tejidos que incluyen: el intestino, el riñón, el hígado, el cerebro y la placenta87. Actúa como un mecanismo de defensa natural frente a diversos fármacos, limitando su absorción intestinal y penetración cerebral, y promoviendo su eliminación en la bilis y la orina. La P-gp se expresa en la membrana apical de todo el intestino, desde el duodeno al recto, con una elevada expresión en los enterocitos del intestino delgado, lo que contribuye a una reducción de la absorción de múltiples fármacos, que son sustratos de este transportador. En los hepatocitos, P-gp se localiza en la membrana canalicular, y en el riñón en el lado luminal de las células epiteliales tubulares proximales, mediando en la eliminación de los xenobióticos en la bilis y en la orina, respectivamente. En la BHE, P-gp es uno de los principales transportadores, localizándose principalmente en la célula endotelial luminal, aunque está presente en otras células incluyendo las neuronas y los astrocitos89. La función de P-gp en la BHE no se comprende bien, pero incluye sustratos con una estructura muy diferente que se fijan a más de un sitio de acción90.
El gen ABCB1 se localiza en el cromosoma7, al igual que el CYP3A4; ambos parecen tener regulaciones y sitios de acción similares y, por tanto, parecen compartir por los mismos sustratos e inhibidores, aunque pueden tener diferente afinidad hacia ellos. Es posible que la P-gp pueda ser también inducida, y que su exceso de expresión pueda originar que el fármaco en cuestión tenga una absorción gastrointestinal limitada, o una reducción de la penetración cerebral, o un incremento de la eliminación por la bilis y la orina. En el intestino, tanto el CYP3A4 como P-gp parecen actuar conjuntamente, disminuyendo la absorción de su sustrato común y contribuyendo a lo que se ha denominado el metabolismo de primer paso (véase la sección sobre los CYP).
Variaciones genéticasLa literatura sobre las UGT está cambiando lentamente al mismo tipo de terminología genética que se usa para los CYP. Sin embargo, los artículos sobre las variaciones genéticas de ABCB1 resultan mucho más confusos. Normalmente se centran en los polimorfismos de un único nucleótido (SNP), o los haplotipos. Un SNP es una variación secuencial de ADN que se produce cuando varía un único nucleótido (A: adenina, T; timina, C: citosina o G: guanina). Un haplotipo es una combinación de alelos en las localizaciones adyacentes de un cromosoma, heredados conjuntamente. La no utilización de los alelos numéricos denota que no está claro cuáles están asociados a las variaciones funcionales en la expresión de P-gp, y cuáles no91. Por ello no es sorprendente que los intentos de completar un metaanálisis del resultado de las asociaciones entre la variaciones genéticas de ABCB1 y los FAE92, o la segunda generación de antipsicóticos, resulten negativos93. La existencia de muchos hallazgos significativos, que raramente se replican, sugiere al autor que las variaciones genéticas de ABCB1 no llegarán a la práctica en los próximos 5años.
Interacciones farmacológicas en generalAlgunos artículos87 clasifican a los sustratos de P-gp en 2 grupos: a)la mayoría de los fármacos, que son sustratos tanto de la P-gp como del CYP3A4, y b)aquellos específicos de la P-gp y que no son metabolizados por los CYP. Los últimos incluyen a unos pocos fármacos tales como dabigatrán (un anticoagulante oral), digoxina y fexofenadina (un antihistamínico). También se cree que no todos los sustratos del CYP3A4 son sustratos de P-gp; midazolam es un sustrato del CYP3A4 pero no de la P-gp94.
La quinidina y la claritromicina han demostrado ser inhibidores de la digoxina, y se cree que esta inhibición está explicada por la potente inhibición de la P-gp94,95. Sin embargo, la quinidina es también un potente inhibidor del CYP2D6, y la claritromicina es un potente inhibidor de CYP3A4. Se cree que otros inhibidores de la P-gp son el verapamilo, la ciclosporinaA, la reserpina, la yohimbina y el tamoxifeno96. Se están intentando desarrollar inhibidores de P-gp altamente selectivos, que no inhiban el CYP3A4 u otros transportadores96.
De igual modo, la rifampicina y el hipérico parecen ser inductores de la P-gp, según han demostrado a través de los efectos de ciertos fármacos tales como la digoxina87,95 y la fexofenadina95, que son sustratos de la P-gp aunque no son metabolizados por el CYP3A4. La complejidad de estudiar los inductores de la P-gp se demuestra por el hecho de que la rifampicina puede ser inicialmente un inhibidor de P-gp e incrementar la absorción de la digoxina, aunque tras cierto tiempo (una semana) puede ser un inductor de P-gp y disminuir su absorción94.
La mejor prueba de que la ciencia sobre los transportadores en general —y sobre la P-gp en particular— puede no estar preparada para la práctica clínica es que la revisión de la información en los prospectos de Estados Unidos y Europa indica a menudo que no existe acuerdo en la información sobre el mismo fármaco en cuanto a la función del transportador, o ni siquiera en su terminología87.
Interacciones farmacológicas en neuropsicofarmacologíaLos artículos sobre la P-gp en la neuropsicofarmacologíason muy confusos para los clínicos. Akamine et al.97 describen a casi todos los antidepresivos y los antipsicóticos como sustratos del P-gp, y reportan que todos ellos pueden inhibirla. Por otro lado, Moons et al.93 sugieren que no todos los antipsicóticos de segunda generación pueden ser sustratos de P-gp, y O’Brien et al.98 reconocen que no queda bien establecido si la inhibición de P-gp por parte de los antidepresivos es clínicamente relevante o no. La confusión de la información se aclara tras la lectura de una revisión de los FAE, donde Zhang et al.99 establecen que: a)los diferentes modelos in vitro de la función de la P-gp dan diferentes resultados en relación al hecho de que los FAE sean o no sustratos, y b)no existe consenso de criterio para definir los sustratos de P-gp entre los FAE. Los artículos tienden a describir a los inductores potentes tales como carbamazepina y fenitoína como inductores de la P-gp. El AVP es normalmente descrito, no como un sustrato, sino como un inductor de P-gp97. La limitada información disponible sugiere que las benzodiazepinas pueden no ser sustratos de la P-gp100,101, sino que pueden ser sustratos de otros transportadores de eflujo localizados en la BHE102.
Para entender mejor nuestras limitaciones actuales a la hora de comprender la interacción farmacológica mediada por la P-gp deberíamos mencionar que Lin103 propuso que los estudios en animales sugieren que la interacción farmacológica al nivel del transporte puede tener mayor impacto en la distribución tisular, particularmente en el cerebro, que en la distribución sistémica medida por las concentraciones plasmáticas. De ser esto cierto, complicaría la interpretación de la interacción farmacológica por parte de los clínicos, ya que el primer paso para interpretar dicha interacción es la medición de los aumentos y las disminuciones en las concentraciones plasmáticas. Si existen se deben asumir un componente farmacocinético pero, de no existir se debe asumir que la interacción es farmacodinámica. Sin Lin tiene razón103 de que las interacciones farmacológicas de la P-gp pueden ser interacciones farmacocinéticas sin influir en la monitorización terapéutica (MT), entonces dichas interacciones farmacocinéticas de la P-gp podrían confundirse con las interacciones farmacodinámicas.
Mecanismo de inducción y receptores nuclearesLa inducción de las enzimas metabólicas, tales como los CYP y las UGT, y de los transportadores, tales como la P-gp, se explica porque la cantidad de dichas proteínas aumenta cuando son inducidas. El incremento de estas proteínas puede lograrse incrementando la síntesis o disminuyendo la degradación. La inducción de muchas enzimas metabólicas y transportadores parece estar mediada principalmente por el incremento de la síntesis. Se conocen 2 excepciones: el CYP2E1 y el CYP2D6. La inducción del CYP2E1 parece estar mediada por un mecanismo completamente diferente al nivel posterior a la transcripción, que origina una estabilización de la proteína que demora su destrucción. El CYP2E1 tiene una vida media corta. Los inductores del CYP2E1, tales como el etanol y la isoniazida, incrementan la vida media de CYP2E1 al disminuir su degradación. Siempre se ha considerado a CYP2D6 como uno de los CYP que no puede inducirse; por tanto, es sorprendente que la actividad del CYP2D6 se incremente durante el embarazo. En un estudio reciente utilizando ratones con CYP2D6 humanizado, Koh et al.24 propusieron que durante el embarazo se produce una disminución de un represor del CYP2D6.
El incremento de la síntesis está a menudo mediado por un grupo de receptores que activan los genes en el núcleo celular, y que se denominan normalmente receptores nucleares. Como dichos receptores nucleares tienen la capacidad de fijarse directamente al ADN y regular la expresión de los genes adyacentes, se les considera factores de transcripción.
Los receptores nucleares constan de 3 grandes dominios proteicos: a)un dominio altamente conservado y vinculado al ADN que une el receptor a las regiones promotoras específicas de los genes diana; b)un dominio menos conservado vinculado al ligando, y c)una zona que reconoce otros factores y co-activadores de la transcripción104. Existe una gran comunicación entre los diferentes receptores nucleares y otros factores de transcripción105. De hecho, uno de los receptores descritos en esta sección, el receptor de aril hidrocarburos (AhR), no es un receptor nuclear sino que forma parte de otra superfamilia de factores de transcripción.
Los receptores nucleares más importantes implicados en la inducción incluyen el receptor de pregnanoX (PXR) y el receptor constitutivo de androstano (CAR). Se cree que el receptor de glucocorticoides (RG)106 y los receptores de estrógenos (RE)107 pueden estar implicados en ciertos fenómenos de inducción. Los RE pueden ser particularmente importantes en la inducción de ciertas enzimas durante el embarazo107.
Algunas familias de receptores nucleares incluyen a los receptores de esteroides (incluyendo RG y RE) y a las hormonas tiroideas que se localizan en el citoplasma, pero que se trasladan al núcleo tras fijarse al sustrato. Existen otros receptores que inicialmente fueron denominados receptores huérfanos, puesto que su función era desconocida108. PXR y CAR se incluyen en esta categoría de receptores nucleares huérfanos; sus clasificaciones oficiales respectivas son NR1I2 y NR1I3109,110.
Uno de los problemas para los clínicos que intentan familiarizarse con esta área de investigación es su complejidad. La función de los receptores nucleares va más allá de los efectos de los xenobióticos tales como los fármacos, pesticidas, contaminantes ambientales, carcinógenos o ciertos nutrientes complejos (como los flavonoides), o la toxicología y la farmacología. Los receptores nucleares participan en la fisiología básica, ya que los muchos compuestos endógenos incluyendo los compuestos biliares, las hormonas y las vitaminas, se fijan a estos receptores nucleares. De hecho, dichos receptores regulan uno de los procesos de biosíntesis más complejos, la transformación del colesterol en múltiples agentes biológicos complejos, con una estructura molecular derivada del colesterol, que incluye a los ácidos biliares, los corticoides, las hormonas sexuales y la vitaminaD. Para complicar aún más la literatura, existen ciertas diferencias importantes entre los receptores nucleares de los humanos y los roedores, lo que complica la extrapolación. Como la literatura sobre estos receptores nucleares está en su infancia, y es extraordinariamente compleja, a menudo describe información contradictoria. En resumen, esta sección aporta algunas pistas acerca del modo en que los inductores de los FAE pueden influir sobre los CYP, pero no se intenta resumir los mecanismosde inducción de la UGT o de la P-gp. Aunque probablemente estos mecanismos están controlados por los mismos receptores nucleares, la información publicada sobre las UGT o la P-gp es tan contradictoria104,111,112 que es imposible hacer un resumen coherente para los clínicos.
AhRAhR se revisa en primer lugar en esta sección puesto que no es realmente un receptor nuclear. En ausencia de un ligando, el AhR se localiza en el citoplasma, pero después de unirse llega al núcleo celular y se convierte en un factor de transcripción. Los AhR están ampliamente distribuidos en diversos tejidos, y son importantes probablemente para el desarrollo embrionario. Ciertos inductores, incluyendo algunos fármacos como el omeprazol, los vegetales crucíferos y los hidrocarburos aromáticos policíclicos (HAP) presentes en el humo, el café tostado y la carne a la parrilla, se unen al AhR, que es importante para la inducción de la familia CYP1, particularmente el CYP1A2 y ciertas UGT.
PXRLos PXR se localizan principalmente en el hígado y el intestino delgado. La carbamazepina, la fenitoína, el fenobarbital, la rifampicina y el hipérico activan los PXR. Estos receptores son importantes para la inducción de las familias CYP2 y CYP3, y ciertas UGT. En un estudio in vitro, Sinz et al.113 exploraron los compuestos que activan los PXR y los clasificaron, conforme a su potencia como inductores del CYP3A4, en 3 grupos (la rifampicina se situaba en el primer grupo de compuestos más activos, la fenitoína y el fenobarbital en el segundo grupo, y la carbamazepina en el tercero con menor potencia).
CAREl CAR se halla únicamente presente en mamíferos, localizándose principalmente en el hígado y el riñón. Se cree que la fenitoína y el fenobarbital activan el CAR, aunque dicho proceso es bastante complejo y no se comprende bien. El CAR es un importante inductor de las familias CYP2 y CYP3, y de la UGT1A1. De acuerdo con Pascussi et al.105, el CAR muestra cierta selectividad pronunciada hacia el CYP2B6 en comparación con el CYP3A4, aunque el PXR regula a ambos sin selectividad.
REDe acuerdo con estudios in vitro: a)los elevados niveles de estrógenos (se multiplican por 100) durante el embarazo activan tanto el RE como el CAR, lo que tiene efectos sinérgicos de incremento de la expresión de CYP2B6107, y b)RE puede ser parcialmente responsable de la inducción de UGT1A4 vista con altos niveles de estrógenos, lo que puede explicar la inducción de la lamotrigina durante el embarazo114.
RGLa inducción del CYP3A4 por parte de los corticoides como la dexametasona, la prednisolona y la metilprednisolona es parcialmente mediada por el RG, aunque el PXR puede ser también importante115.
Variaciones genéticasSe han publicado intentos preliminare explorando el modo en que las variaciones genéticas del AhR, el PXR y/o el CAR pueden explicar las diferencias entre las personas en la respuesta a los inductores109. La dificultad de interpretar dichos estudios es que todos los artículos de revisión sobre la materia insisten en que los receptores nucleares y los AhR parecen actuar de modo coordinado, con gran superposición entre dichos factores de transcripción, y con amplias acciones en los CYP, las UGT y los transportadores. Por tanto, para estudiar el modo en que las variaciones genéticas influyen sobre la inducción de un fármaco que, por ejemplo, está principalmente metabolizado por el CYP1A2, se deben estudiar todas las variaciones genéticas al mismo tiempo (del CYP1A2, del AhR, del PXR y del CAR).
La compleja farmacocinética de la polifarmacia puede requerir la modificación de los factores de correcciónEste artículo incluye factores de corrección en la tabla 3, con idea de ayudar a corregir los efectos inductivos en la neuropsicofarmacología, lo que permitirá a continuación la modificación de las dosificaciones de los fármacos para aproximarse al efecto de los inductores en un paciente específico. Sin embargo, la literatura advierte que la extrapolación a un sujeto promedio es problemática, porque existe gran variabilidad de los efectos de los inductores en la población116. Lamentablemente, es preciso pero los factores de corrección son una mera aproximación, pero es mejor disponer de ellos que no tenerlos.
Esta quinta sección revisa 4 situaciones que pueden modificar los efectos inductivos: la vía no oral, los individuos ML, la presencia de inhibidores y la presencia de otros inductores.
Las vías no orales, en particular la vía intravenosa, están menos influenciadas por la inducción que la vía oral117. Las personas que carecen de una isoenzima activa, tales como los ML del CYP2D6 o del CYP2C19, no pueden ser inducidos en ese CYP del que carecen, pero otros CYP pueden ser inducidos. La risperidona es metabolizada principalmente a nivel del CYP2D6, pero su metabolismo se ve alterado en los ML a nivel de otros CYP2D6, quienes tienen aproximadamente la mitad de capacidad para metabolizar la risperidona118 y necesitan solamente la mitad de la dosis promedio119. Los inductores de los CYP no pueden inducir el CYP2D6, pero sí el CYP3A4, que es una enzima metabólica auxiliar del metabolismo de la risperidona. De hecho, en un paciente promedio de risperidona que tome inductores potentes del CYP3A4, esta isoenzima se convierte en la enzima metabólica más importante para el metabolismo de la risperidona, exigiendo que se duplique la dosis de risperidona en esa persona119. Un factor de corrección de 2 indica que la toma de un inductor potente del CYP3A4 (factor de corrección=2) en un ML del CYP2D6 (factor de corrección=0,5) se cancelan mutuamente contribuyendo a una eliminación cercana a la normal. En la realidad, los MP del CYP2D6 que toman inductores potentes del CYP3A4 tienen un metabolismo de la risperidona algo inferior al normal119.
Es difícil predecir los resultados en situaciones donde se combinan inductores e inhibidores. En el caso de la risperidona, los inductores de CYP3A4 tienen efectos más potentes que un inhibidor potente de la risperidona tal como la fluoxetina, que inhibe a ambas enzimas metabólicas de la risperidona119. En esta situación de combinación de inhibidores e inductores es mejor utilizar la MT para establecer el resultado. El nivel de inducción de la tabla 5, medido con el número de «+», puede ayudar a los clínicos. Los efectos de los inductores potentes del CYP3A4 y del CYP2B6 es masivo («4+» en la tabla 5), y mucho más importantes que los efectos de los inhibidores. La inducción de CYP1A2 es probablemente solo moderada («2+»en la tabla 5) y, conforme a nuestra experiencia con los inductores potentes de la clozapina, tiende a tener menos potencia que los inhibidores potentes tales como la fluvoxamina120. Por otro lado, la inducción del CYP2C9 y del CYP2C19 es mínima («1+» en la tabla 5) y claramente menor que el efecto de los inhibidores potentes. La fenitoína es metabolizada por el CYP2C9 y el CYP2C19, y es un inductor de su propio metabolismo, aunque estos efectos inductivos probablemente no tienen relevancia clínica. La fenitoína en concentraciones elevadas es un inhibidor potente del CYP2C9 y del CYP2C19, lo que es clínicamente relevante durante la intoxicación con fenitoína. En el caso de los fármacos metabolizados por las UGT, como la lamotrigina, los inductores potentes de los FAE requieren una duplicación de la dosis, mientras que el valproato, un inhibidor potente, requiere reducir la dosis a la mitad. La combinación de inductores potentes como la fenitoína y el valproato requiere el uso de dosis normales de lamotrigina. En esta situación puede ser más inteligente utilizar la MT de la lamotrigina, puesto que los efectos inhibitorios del valproato pueden ser más fuertes que los de los inductores74.
La literatura da pocas recomendaciones sobre el modo de predecir lo que ocurre con las combinaciones de los inductores. El autor tiene experiencia en 2 tipos de situaciones utilizando combinaciones de: a)FAE con potentes propiedades inductoras con otros inductores del CYP1A2, y b)FAE con potentes propiedades inductoras con FAE inductores leves del CYP3A4. Cuando los pacientes toman fármacos metabolizados por CYP1A2, el tabaquismo parece influir en la inducción por parte de AhR, mientras que la inducción por parte de los FAE es mediada por PXR y/o CAR. Por tanto, el hábito de fumar y los inductores de los FAE parecen tener efectos inductores aditivos e independientes120.
Cuando los pacientes toman fármacos metabolizados por el CYP3A4, el efecto de los inductores potentes tales como la carbamazepina, la fenitoína o el fenobarbital es mucho más potente que el de los inductores leves de CYP3A4 tales como clobazam, eslicarbazepina, oxcarbazepina, rufinamida o topiramato (tabla 4). En estos casos, la administración de un inductor leve cuando un paciente toma un inductor potente puede no tener efecto alguno. Por contra, el intercambio de un inductor potente tal como carbamazepina por oxcarbazepina suele asociar con una disminución de los efectos inductivos (aumentando los niveles del sustrato del CYP3A4), y en cambio el pasar de un inductor leve, tal como la oxcarbazepina, a la carbamazepina se asocia a un incremento importante de los efectos inductivos (disminuyendo los niveles del sustrato de CYP3A4). No existen muchas comparaciones en la literatura acerca de la intensidad de los inductores de CYP3A4, aunque Ohno et al.121, tras revisar la literatura utilizando un complejo modelo matemático, proporcionan algunas comparaciones de los efectos de los diversos inductores del CYP3A4. Ellos describieron incrementos de 7,7 en el metabolismo por parte de la rifampicina (450-600mg/día), 4,7 por parte de la fenitoína (300-400mg/día), 3,0 por parte de la carbamazepina (200-600mg/día), 1,4 por parte de efavirenz, un agente antirretroviral (600mg/día), y 1,2 por parte del hipérico (600-900mg/día).
Las interacciones farmacológicas de tipo farmacodinámico pueden contribuir también a la modificación de los efectos inductoresLos factores de corrección (tabla 3) ayudan a los clínicos a interpretar el efecto de la adición de inductores potentes a un fármaco. La sección anterior trata de aportar a los clínicos una primera visión de la complejidad al explicar que la ruta de administración, el ser un ML, o la prescripción conjunta de inhibidores u otros inductores, influyen en los efectos inductores de los FAE. Esta sección trata de centrarse en la complejidad añadida de la prescripción en el mundo real, describiendo como ejemplos las combinaciones en pares de 3 FAE importantes: carbamazepina, fenitoína y AVP, carbamazepina-AVP, carbamazepina-fenitoína y fenitoína-AVP. El autor se ha encontrado con cientos de pacientes epilépticos y/o psiquiátricos que toman estas combinaciones, y ha visto que normalmente los neurólogos y/o psiquiatras que tratan a estos pacientes no tienen ni idea de la complejidad de estas interacciones farmacológicas, probablemente las interacciones más complicadas en neuropsicofarmacología, y las más inciertas en cuanto a su resultado.
La partei describe: a)a la fenitoína como un inductor potente de múltiples enzimas, que en concentraciones plasmáticas elevadas puede saturar al CYP2C9 y al CYP2C9, y b)a muchos de los fármacos denominados inductores leves en este artículo, incluyendo el VPA, como fármacos que pueden ser inhibidores con relevancia clínica. Por tanto, el primer nivel de complejidad es que a menudo los fármacos pueden ser tanto inductores como inhibidores. La tabla 8 trata de aportar una visión sobre la dificultad en la interpretación de estas interacciones farmacológicas, puesto que pueden combinar componentes complejos tanto farmacodinámicos como farmacocinéticos. Para manejar las interacciones farmacológicas de estos 3 FAE se deben comprender otros niveles de complejidad, sabiendo que la unión a las proteínas plasmáticas es muy importante para la fenitoína y el AVP, y a veces posiblemente relevante para la carbamazepina. Otro nivel de complejidad es que los mecanismos farmacodinámicos son también importantes para interpretar estas interacciones farmacológicas. Como este artículo de revisión se centra en la inducción, no puede revisar detalladamente las uniones proteicas o la farmacodinámica, aunque la tabla 8 proporciona una amplia descripción de los complejos mecanismos farmacológicos de estas 3 interacciones farmacológicas. Otras publicaciones previas aportan una información más amplia, incluyendo los mecanismos farmacodinámicos y las uniones proteicas74,122,123. En resumen, el autor propone que los psiquiatras y neurólogos no deberían utilizar estas 3 combinaciones a menos que comprendan a fondo los mecanismos farmacológicos que están detrás de las interacciones farmacológicas y hagan uso de la MT.
Combinaciones de los FAE con gran complejidad farmacocinética y farmacodinámica
| Carbamazepina y AVP |
| Resumen para los clínicos: tengan cuidado y utilicen la MT, incluyendo las concentraciones libres |
| Efectos farmacocinéticos |
| Efectos del AVP sobre las concentraciones séricas de la carbamazepina: ↑ totales y libres |
| (−) epóxido hidroxilasa: ↑ concentración sérica del epóxido |
| (−) UGT2B7: ↓ concentración sérica del metabolito diol glucuronizado |
| ↓ la carbamazepina unida a las proteínas plasmáticas |
| Efectos de la carbamazepina en las concentraciones séricas del AVP: ↓ totales y ↑ libres |
| Probable inhibición de algunas enzimas metabólicas |
| ↓ el AVP unido a las proteínas plasmáticas |
| Efectos farmacodinámicos (no se entienden bien) |
| La carbamazepina bloquea los canales del sodio activados por voltaje y el AVP tiene efectos anticonvulsivos complejos |
| Posibles efectos aditivos en la estabilización del estado de ánimo al actuar en el sistema de señalización intracelular |
| Los libros de texto reportan normalmente un incremento del riesgo de RAM neurológicas |
| Carbamazepina y fenitoína |
| Resumen para los clínicos: tengan cuidado y utilicen la MT, incluyendo las concentraciones libresFarmacocinética |
| Efectos de la fenitoína sobre las concentraciones séricas de la carbamazepina: ↓ totales y ↑ libres |
| Inducción enzimática adicional |
| ↓ la carbamazepina unida a las proteínas plasmáticas |
| Efectos de la carbamazepina sobre las concentraciones séricas de la fenitoína: dificiles de predecir |
| Posible inducción enzimática adicional |
| ↓ la fenitoína unida a las proteínas plasmáticas |
| Posible inhibición del CYP2C19 |
| Farmacodinámica (no se entienden bien) |
| Posibles efectos anticonvulsivos aditivos y ↑ riesgo de RAM: ambos bloquean los canales del sodio activados por voltaje |
| Fenitoína y AVP |
| Resumen para los clínicos: tengan cuidado y utilicen la MT, incluyendo las concentraciones libresFarmacocinética |
| Efectos de la fenitoína sobre las concentraciones séricas del AVP: ↓ totales y ↑ libres |
| Inducción enzimática |
| ↓ el AVP unido a las proteínas plasmáticas |
| Efectos del AVP sobre las concentraciones séricas de fenitoína: ↑ totales y libres |
| Inhibición potente del CYP2C9 |
| ↓ la fenitoína unida a las proteínas plasmáticas |
| Farmacodinámica (no se entienden bien) |
| La fenitoína bloquea los canales del sodio activados por voltaje y el AVP tiene efectos anticonvulsivos complejos |
(−): inhibición; FAE: fármacos antiepilépticos; RAM: reacción adversa a medicamentos; MT: monotorización terapéutica; AVP: ácido valproico.
Durante los últimos 15años como clínico y asesor de pacientes difíciles en neuropsicofarmacología, el autor se ha encontrado con un pequeño número de pacientes con perfiles farmacocinéticos extremadamente raros que parecen ser altamente sensibles a los efectos inductivos, y precisan de dosis masivas de ciertos fármacos para lograr concentraciones séricas terapéuticas. Tras muchos años de consideración, el autor cree que el mejor modo de comprenderlos sería decir que son candidatos a tener perfiles genéticos inusuales a nivel de los receptores nucleares, lo que les hace extremadamente sensibles a los efectos inductivos en las diferentes rutas metabólicas.
La tabla 9124–128 incluye a 6 pacientes identificados por el autor como posibles poseedores de una respuesta excesiva a los inductores, por lo que se realizó un seguimiento de los mismos. La prevalencia de estas 6 personas raras se desconoce, aunque el autor calcula que representan entre 1/1.000 y 1/100 de los pacientes con enfermedades mentales graves. Dos de los 6 pacientes necesitaron dosis muy elevadas de los sustratos de CYP3A4 en presencia de inductores potentes de los FAE (casos1 y 3). Un paciente aparentó ser muy sensible a la inducción de las UGT y precisó dosis de lamotrigina y lorazepam extremadamente elevadas (caso5). Cuatro de los 6 pacientes parecieron tener grandes efectos inductivos secundarios al AVP, demostrados por los efectos en la MT de dicha droga (casos1 y 4), de clozapina (caso2) y olanzapina (caso3). El caso más sorprendente sobre el AVP en el que el paciente desarrolló un carcinoma renal metastásico en el contexto de una esclerosis tuberosa que requirió >10g/día de divalproex sódico para lograr concentraciones séricas terapéuticas del AVP, al administrarse conjuntamente con la fenitoína (caso6). Tres UGT (UGT1A6, UGT1A9 y UGT2B7) están presentes a concentraciones elevadas en los riñones71. Como se describió en la partei, UGT1A6, UGT1A9 y UGT2B7 están probablemente implicadas en el metabolismo del VPA74.
Listado de pacientes con sensibilidad extrema a los efectos inductivos
| Caso 1. Paciente ♂ caucásico hospitalizado con esquizofrenia y epilepsia de inicio tardíoa |
| A)Sensibilidad extrema a la inducción de CYP3A4 |
| -Carbamazepina: anteriormente fue necesario administrar 1.500-2.000mg/díab para alcanzar las concentraciones séricas terapéuticas |
| -Quetiapina |
| Con fenitoína y AVP, el ratio C/D de quetiapina fue 10 veces menor a lo esperado |
| Con AVP, el ratio C/D de quetiapina fue 2-6 veces inferior a lo esperado |
| -Diazepamc |
| Con fenitoína, las concentraciones séricas fueron indetectables a pesar de tomar 30mg/día |
| Con AVP, la eliminación de diazepam tras 30mgi.m. fue ≥3 superior a lo esperado |
| B)Posible autoinducción del AVP al tomarlo como un concentrado, aunque no ocurrió tomando divalproex sódico |
| Con concentrado, se necesitaron 5.250mg/díad para alcanzar las concentraciones terapéuticas |
| Con divalproex sódico fueron suficientes 2.000mg/díae para lograr concentraciones terapéuticas |
| C)El metabolismo de clozapina y olanzapina (fármacos CYP1A2) fue normal para un varón fumadorf |
| Caso 2. Paciente ♂ afroamericano hospitalizado con trastorno esquizoafectivog |
| A)Inducción extrema de la clozapina por el AVP (divalproex sódico) |
| -Fumador (20 cigarrillos/día): se precisó administrar ≥650mg/día para alcanzar concentraciones séricas >350ng/mlh |
| Durante la inducción del tabaco, el paciente demostró una elevada capacidad metabólica para la clozapina |
| -Fumador y AVP: se precisó administrar ∼1.200mg/día para alcanzar concentraciones séricas >350ng/mli |
| La adición de la inducción del AVP originó la mayor capacidad metabólica de la clozapina que el autor haya visto nunca |
| Caso 3. Paciente ♂ caucásico hospitalizado con trastorno esquizoafectivoj |
| A)Sensibilidad extrema a la inducción de CYP3A4 |
| -Carbamazepina: se necesitaron hasta 2.800mg/díab para alcanzar concentraciones séricas terapéuticas; esto se explica parcialmente por los depósitos de tejido graso y el alto volumen de distribuciónk |
| -Risperidona |
| Con carbamazepina, aproximadamente 4 veces superior a la capacidad metabólica normal |
| Con divalproex sódico el metabolismo de la risperidona era normal |
| -Paliperidona: con carbamazepina, alta capacidad de metabolizar la paliperidona |
| B)Inducción extrema de olanzapina por AVP (divalproex sódico) y omeprazol |
| -Olanzapina: con divalproex sódico y omeprazol, 1,5-2 veces superior al metabolismo normal |
| Caso 4. Paciente ♂ caucásico hospitalizado con trastorno bipolarl |
| A)Posible autoinducción de AVP al administrarse divalproex sódico |
| -Dado de alta con 4.000mg/díam |
| Caso 5. Paciente ♂ hospitalizado con epilepsia en el lóbulo frontal y temporal y psicosisn |
| A)Sensibilidad extrema a la inducción de UGT durante el tratamiento con fenitoína y fenobarbital |
| -Lamotrigina: se precisó una dosis 2,6 veces superior a la recomendadao |
| -Lorazepam: se toleraron dosis >20mg/día sin sedación |
| Caso 6. Paciente ♂ hospitalizado con esclerosis tuberosa y tumores renalesp |
| A)Incremento de la capacidad metabólica de AVP al administrarse fenitoína y AVP |
| -Divalproex sódico: incremento de la dosis >10.000mg/día para alcanzar las concentraciones séricas terapéuticasq |
| -Fenitoína: sin cambios en el metabolismo de la fenitoína y sin necesidad de modificar la dosificación |
Dosis habituales de carbamazepina para alcanzar las concentraciones terapéuticas: 800-1.200mg/día. Dosis máxima recomendada: 1.600mg/día124.
El diazepam se metaboliza principalmente por CYP2C19; CYP3A4 es una enzima auxiliar. En este paciente, CYP3A4 era probablemente la enzima metabólica principal para el diazepam.
Los datos fueron publicados sin explicación farmacológica125. El ratio C/D de AVP fue de 0,013-0,017.
Los datos fueron publicados sin explicación farmacológica125. El ratio C/D de AVP fue de 0,036-0,048.
En el momento de la dosis máxima de carbamazepina el IMC era de 40, con un peso corporal de 191kg. La elevada dosis se debe parcialmente a la obesidad128.
El ratio C/D de AVP fue de 0,024-0,033 durante el primer mes, disminuyendo a 0,017-0,018 durante el segundo mes.
El paciente necesitó 1.600mg/día para alcanzar las concentraciones séricas terapéuticas de lamotrigina. La dosis máxima recomendada es de 600mg/día.
Seguimiento durante 4años (edad de 44 a 48años hasta que falleció). Inicialmente tenía angiomiolipomas en ambos riñones. Durante el segundo año, una masa creciente en el riñón derecho condujo al diagnóstico de un posible carcinoma renal, y el resultado patológico de la nefrectomía sugirió un angiomiolipoma. Al tercer año,aparecieron metástasis cerebrales.
Al principio, cuando el paciente tenía tumores renales bilaterales, precisó cerca de 5.000mg/día de divalproex sódico para alcanzar las concentraciones terapéuticas, con ratios C/D de AVP de 0,010-0,018. Tras la nefrectomía, una vez que la presencia del cáncer renal metastásico era obvia, precisó 10.500mg/día de divalproex para alcanzar las concentraciones terapéuticas, con ratios C/D de AVP de 0,005-0,009.
Este artículo, dividido en 2 partes, aporta información a los clínicos para compensar la falta de atención en la literatura acerca de las propiedades inductivas de los FAE. La partei introduce la cuestión de los efectos inductores potentes (tabla 1) y leves (tabla 2), incluyendo recomendaciones clínicas para corregir el efecto de los inductores potentes mediante modificaciones de las dosis de los sustratos inducidos, utilizando factores de corrección (tabla 3).
La parteii trata de educar a los clínicos acerca de la complejidad de la interpretación de las interacciones farmacológicas de los FAE, aportando un conocimiento farmacológico básico para ayudar a mejorar su capacidad de interpretar las interacciones farmacológicas complejas.
Se revisa la farmacología básica de los CYP, las UGT, la P-gp y los receptores nucleares. Seis isoenzimas CYP (el CYP1A2, el CYP2B6, el CYP2C9, el CYP2C19, el CYP2D6 y el CYP3A4) son definitivamente importantes para el metabolismo de los fármacos neuropsicofarmacológicos. El CYP2B6 y el CYP3A4 son muy sensibles a la inducción. El CYP1A2 es moderadamente sensible, mientras que CYP2C9 y CYP2C19 son solo levemente sensibles. Por último, el CYP2D6 no puede ser inducida por los fármacos (tabla 5). Las variaciones genéticas del CYP2C9, el CYP2C19 y el CYP2D6 son suficientemente conocidas para su uso clínico. Los ML son personas que carecen de las CYP activas específicas; a pesar de que han sido identificadas para el CYP2C19 y el CYP2D6, sus frecuencias varían con la raza (tabla 6).
Las UGT son las enzimas de conjugación más importantes (tradicionalmente denominadas enzimas metabólicas de faseii). Constituyen las enzimas metabólicas principales para algunos antipsicóticos, algunos FAE y algunas benzodiazepinas (tabla 7). La literatura ha prestado menos atención a las UGT, siendo menos conocidas que los CYP60. Entre las UGT importantes para la neuropsicofarmacología están la UGT1A1, la UGT1A3, la UGT1A4, la UGT2B7 y la UGT2B15 (tabla 7). No existe información definitiva acerca del modo en que los inductores influyen en su actividad. De igual modo, no está claro el modo en que las variaciones genéticas o los inhibidores (como el AVP) influyen sobre cualquiera de estas UGT específicas.
Otro gran grupo de proteínas farmacocinéticas, llamadas transportadores, pueden ser también inducidas. Normalmente actúan en conjunción con las enzimas metabólicas para eliminar los xenobióticos del cuerpo. La P-gp constituye el transportador más importante, siendo un componente fundamental de la BHE. Nuestro limitado conocimiento de la P-gp no puede extrapolarse la práctica clínica. Los prospectos de Estados Unidos y Europa no están de acuerdo en la relevancia de los transportadores ni en la terminología utilizada para describirlos. Además, la literatura no llega a un acuerdo acerca de qué fármacos neuropsicofarmacológicos son sustratos, inhibidores o inductores de la P-gp.
Al crecer rápidamente la literatura sobre los receptores nucleares está comenzando a aportar cierto entendimiento de los mecanismos de los inductores. La inducción de las enzimas metabólicas tales como los CYP y las UGT, y los transportadores tales como la P-gp, implica que la cantidad de dichas proteínas se incrementa durante la inducción. Dicho incremento se explica casi siempre mediante el incremento de la síntesis mediada por un grupo de receptores que activan los genes en el núcleo celular, denominados receptores nucleares. Dichos receptores nucleares son considerados factores de transcripción, ya que pueden unirse directamente al ADN y regular la expresión de los genes adyacentes. Los receptores nucleares más importantes implicados en la inducción son el PXR y el CAR. Los receptores hormonales RG y RE pueden tener también cierto papel en la inducción. AhR no es un receptor nuclear, aunque forma parte de otra superfamilia de factores de transcripción, y media en los efectos inductivos del humo del tabaco sobre el CYP1A2. La literatura ha comenzado a explorar el modo en que las variaciones genéticas en el AhR, el PXR y/o el CAR pueden explicar las diferencias entre las personas,a la respuesta a los inductores. Los receptores nucleares y el AhR parecen actuar de modo coordinado, con gran superposición de estos factores de transcripción, y con acciones amplias en los CYP, las UGT y los transportadores. Por tanto, no será fácil establecer a nivel de la inducción de las variaciones genéticas específicas a nivel de genes individuales.
La tabla 3 incluye los factores de corrección para los FAE con propiedades inductivas potentes. Sin embargo, la literatura advierte que la extrapolación a las personas es problemática, dado que existe gran variabilidad de los efectos de los inductores en la población. Las vías no orales, en particular la vía intravenosa, se ven menos influidas por la inducción que la vía oral. Los ML no pueden ser inducidos en el CYP que carecen, aunque pueden ser inducidos en otros CYP. Es difícil predecir los resultados cuando existen combinaciones de inductores e inhibidores. La literatura proporciona muy pocas recomendaciones acerca del modo de explicar lo que sucede con las combinaciones de inductores. El autor tiene experiencia con 2 tipos de situaciones que utilizan las combinaciones de: a)FAE con potentes propiedades inductoras con otros inductores del CYP1A2, y b)FAE con potentes propiedades inductoras con FAE inductores leves del CYP3A4. Cuando los pacientes toman fármacos metabolizados por el CYP1A2, el hábito de fumar parece influir en la inducción a través del AhR, mientras que la inducción por parte de los FAE es mediada por el PXR y/o el CAR. Por tanto, el tabaquismo y los inductores de los FAE parecen tener efectos aditivos e independientes. Cuando los pacientes toman fármacos metabolizados por el CYP3A4, el efecto de los inductores potentes tales como la carbamazepina, la fenitoína o el fenobarbital es mucho más potente que el de los inductores leves del CYP3A4 tales como el clobazam, la eslicarbazepina, la oxcarbazepina, la rufinamida o el topiramato (tabla 4). En dichos casos, la administración de un inductor leve cuando el paciente toma un inductor potente puede no tener efecto alguno.
La tabla 8 utiliza como ejemplo las combinaciones de carbamazepina-AVP, carbamazepina-fenitoína y fenitoína-AVP para demostrar la complejidad de ciertas interacciones farmacológicas de los FAE. Para comprender el resultado de estos 3 pares de combinaciones no solo es necesario tener en cuenta los efectos inhibitorios e inductivos, sino las interacciones farmacológicas de tipo farmacodinámico y la unión a las proteínas plasmáticas. Otras publicaciones previas dan más información, incluyendo los mecanismos farmacodinámicos y la unión a las proteínas plasmáticas, para comprender las interacciones farmacológicas en neuropsicofarmacología74,122,123.
Durante los últimos 15años como clínico y asesor de pacientes difíciles en neuropsicofarmacología el autor se ha encontrado con un pequeño número de pacientes con perfiles farmacocinéticos extremadamente raros, probablemente debido a sus perfiles genéticos raros a nivel de los receptores nucleares, que les hace altamente sensibles a los efectos inductivos de los FAE en diferentes rutas metabólicas. La tabla 9 describe a 6 pacientes, incluyendo: a)2 que precisaron altas dosis de sustratos del CYP3A4 en presencia de FAE con potentes propiedades inductoras; b)4 con grandes efectos inductivos secundarios al AVP; c)uno muy sensible a la inducción de las UGT, requiriendo dosis extremadamente elevadas de lamotrigina y lorazepam, y d)un paciente con esclerosis tuberosa que precisó tomar >10g/día de divalproex sódico para lograr las concentraciones séricas terapéuticas de AVP, al tomarlo en combinación con la fenitoína.
Conflicto de interesesNinguna organización comercial ha participado en la escritura de este documento, para su publicación. El autor reporta la ausencia de relación financiera, con intereses comerciales, durante los últimos 36 meses.
El autor reconoce la labor de Lorraine Maw, M.A., y Margaret T. Boden, R.N., M.L.T., del Centro de Investigación en Salud Mental en el Eastern State Hospital, Lexington, KY, Estados Unidos, por su colaboración para la edición de este artículo.