




La enfermedad de Creutzfeldt-Jakob (ECJ) es una patología neurodegenerativa perteneciente al grupo de las enfermedades priónicas o encefalopatías espongiformes transmisibles. Está causada por el depósito en el sistema nervioso central de una isoforma patológica de la proteína priónica normal (PrPc) presente en todos los mamíferos. El mecanismo por el que se produce esta alteración conformacional se desconoce. El acúmulo de la proteína priónica patológica (PrPsc) da lugar a una degeneración neural que provoca un deterioro neurológico rápidamente progresivo de pronóstico fatal.
La ECJ se clasifica en las formas esporádica (ECJe), familiar y adquirida. La forma esporádica representa el 85% de los casos con una mayor incidencia en torno a los 60 años y el 90% de los pacientes fallecen en un año desde la instauración de los síntomas con una supervivencia media de 6 meses1.
Las manifestaciones clínicas clásicas de la ECJe son la demencia rápidamente progresiva, mioclonías, signos piramidales, extrapiramidales y cerebelosos. Aunque con menor frecuencia, la enfermedad puede comenzar con clínica psiquiátrica inespecífica como cambios en la personalidad, alteración del comportamiento, ansiedad, depresión e incluso como un cuadro psicótico lo que puede dificultar el diagnóstico inicial2-11.
El diagnóstico se basa en la clínica y los hallazgos de la exploración neurológica junto a la presencia de alteraciones en las secuencias de difusión (DWI) o FLAIR en la RMN cerebral en el núcleo caudado y putamen o en al menos 2 regiones corticales12, un registro EEG que muestre complejos periódicos de ondas agudas sobre actividad de fondo lentificada13, y/o la positividad de la proteína 14-3-3 en LCR1. No obstante, estos hallazgos no son patognomónicos y su normalidad no descarta la enfermedad. El diagnóstico definitivo se establece mediante el análisis anatomopatológico que demuestre degeneración espongiforme, pérdida neuronal y gliosis1. En la actualidad no existe una terapia curativa, siendo el tratamiento meramente sintomático.
Presentamos el caso de una mujer colombiana de 53 años divorciada con un hijo de 20 años. Sin otros familiares directos y muy reducida red social de apoyo. Sin antecedentes psiquiátricos personales ni familiares. Comenzó con ideación delirante de perjuicio que precisó ingreso involuntario en la Unidad de Agudos de Psiquiatría. Se realizó análisis sanguíneo, tóxicos en orina y TC craneal sin anomalías. Durante la hospitalización se instauró tratamiento con risperidona hasta 6mg/día con buena respuesta. Se dio de alta con el diagnóstico de trastorno psicótico no especificado y se mantuvo tratamiento con paliperidona 9mg/24h.
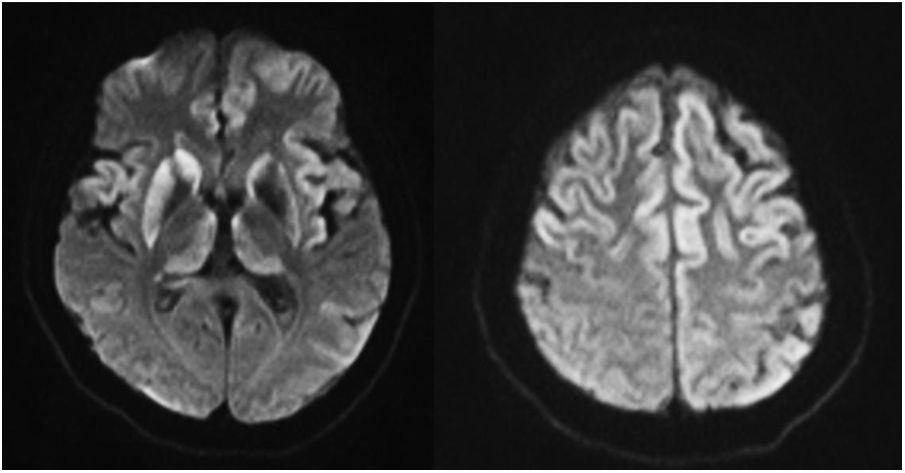
Durante el seguimiento ambulatorio en consultas externas de salud mental a los 8 meses se apreció reaparición del trastorno delirante considerándose secundario a una mala adherencia terapéutica, requiriendo un nuevo ingreso en Psiquiatría. En esta ocasión no se logró control farmacológico completo de la clínica psicótica y se detectó además un enlentecimiento en la marcha y déficits cognitivos. Ante la ausencia de soporte familiar, al alta se trasladó a un centro sociosanitario de media estancia y se solicitó valoración en consultas externas de Neurología. La RM cerebral ambulatoria mostró hiperintensidad en T2 y FLAIR con restricción de la difusión del córtex frontal e insular, de los tálamos y de los ganglios basales, de predominio derecho (fig. 1). El EEG registró un enlentecimiento global de carácter encefalopático moderado.
 y tálamos (núcleo dorsomedial).")
En este momento la paciente presentaba severa desorientación temporal y espacial, limitaciones en la deambulación, piramidalismo generalizado y mioclonías. A nivel funcional, pérdida completa de la autonomía personal, no emitía lenguaje y estaba confinada en una silla de ruedas. La paciente presentaba importantes alteraciones de la conducta y no se pudo realizar con éxito la punción lumbar.
Dado que nuestra paciente presentó un deterioro cognitivo rápidamente progresivo, signos extrapiramidales, piramidalismo y mutismo acinético, con una RM cerebral compatible, fue diagnosticada de ECJe probable, según los criterios diagnósticos de consenso internacional1.
Cabe destacar que, aunque se consideran infrecuentes, los síntomas psiquiátricos se encuentran presentes en la ECJe como síntomas prodrómicos y en estadios precoces de la enfermedad hasta en un 40% de casos14; sin embargo, la presentación en forma de un cuadro psicótico puro durante tantos meses de evolución es atípica y las alteraciones de predominio frontal en las secuencias de difusión en la RNM de nuestra paciente indican correlación con la sintomatología de inicio.
En resumen, nuestro caso ilustra la necesidad de considerar como diagnóstico diferencial la ECJ en pacientes con clínica psicótica o trastornos afectivos refractarios al tratamiento psiquiátrico convencional. La falta de marcadores biológicos para el diagnóstico de enfermedades psiquiátricas en muchas ocasiones puede dificultar el diagnóstico inicial, especialmente en casos de presentación atípica15,16.
Por tanto, en pacientes en los que haya un alto índice de sospecha de ECJ inicial con EEG y prueba de neuroimagen normales, se recomienda repetir estas pruebas durante el curso de la enfermedad14.