



El desarreglo del desarrollo sexual es la anormalidad e incongruencia entre el sexo cromosómico, el sexo gonadal y el fenotipo. El hermafroditismo verdadero constituye menos del 10% de los desarreglos del desarrollo sexual. En este artículo se reporta el caso de un hermafroditismo verdadero que cursa con un teratoma quístico.
Caso clínicoPaciente fenotípicamente masculino, de 53 años de edad. Consultó por cuadro de dolor abdominal. Las imágenes radiológicas evidencian masa abdominal quística compleja en hemiabdomen izquierdo. Es llevado a laparotomía donde se encuentra masa con pedículo torcido correspondiente a trompa de Falopio y ovario izquierdo, se aprecian órganos genitales internos femeninos (ovarios, trompas de Falopio y útero). Reporte de patología: encuentra salpingooforectomía izquierda compatible con teratoma quístico maduro con necrosis hemorrágica y trompa de Falopio con necrosis hemorrágica, útero (cervicitis crónica leve, endometrio atrófico) y ovoteste derecho. El cariotipo fue reportado como 46XX.
DiscusiónLos pacientes con hermafroditismo verdadero tienen un 2,6-4,6% de riesgo de desarrollar una neoplasia gonadal, el caso del paciente mencionad comienza con abdomen agudo secundario a masa ovárica torcida.
ConclusionesSe presenta el caso de un paciente adulto fenotípicamente masculino, al cual se le diagnostica hermafroditismo verdadero, cariotipo 46XX, quien presento torsión de un teratoma quístico maduro izquierdo.
The disorder of sex development is the abnormality or inconsistency between chromosomal sex, gonadal sex, and phenotype. True hermaphroditism is found in less than 10% of the disorders of sex development. In this article, a case is reported on a true hermaphroditism that caused a cystic teratoma.
Case reportA 53 year-old phenotypical male patient consulted for abdominal pain. The radiological images showed a complex cystic abdominal mass in left side of the abdomen. A laparotomy was performed that found a pedunculated mass corresponding to a twisted fallopian tube and left ovary, as well as observing internal female genital organs (ovaries, fallopian tubes and uterus). Pathology reported a left salpingo-oophorectomy compatible with a mature cystic teratoma and fallopian tube with haemorrhagic necrosis, a uterus (mild chronic cervicitis, atrophic endometrium) and right ovotestis. The karyotype was reported as 46XX.
DiscussionPatients with true hermaphroditism have a 2.6-4.6% risk of developing gonadal neoplasia. In the patient reported, it debuted with acute abdomen secondary to a twisted ovarian mass.
ConclusionsThis is a case of a phenotypical male adult patient, who was diagnosed with true hermaphroditism, 46XX karyotype, and presented with left twisted mature cystic teratoma.
Se define como desarreglo o trastorno del desarrollo sexual (DDS) a la anormalidad e incongruencia entre el sexo cromosómico, el sexo gonadal y el fenotipo. Este término viene reemplazando al de intersexualidad, y en general se refiere a trastornos de la diferenciación sexual que con frecuencia dan una ambigua apariencia de los genitales externos1,2. Pueden ser, según la clasificación tradicional: seudohermafrodita femenino (con presencia de 2 ovarios), seudohermafrodita masculino (con 2 testículos), hermafrodita verdadero (tejido testicular y ovárico en el mismo paciente), disgenesia gonadal (gónada rudimentaria –ni testículo ni ovario–) y pura (si ambas gónadas son rudimentarias).
Los casos de hermafroditismo verdadero constituyen menos del 10% de los DDS, presentándose en uno de cada 20.000 recién nacidos y se conocen más de 400 casos reportados en la literatura. Aunque se presentan casos familiares, la mayoría son esporádicos. Hermafroditismo verdadero se define como la presencia de tejido ovárico (contiene folículo de Graaf) y tejido testicular (con los distintos túbulos) en el mismo individuo. Ambas estructuras, de Müller y de Wolff, pueden estar presentes y los genitales externos son generalmente atípicos, aunque se han identificado casos en hombres fenotípicamente normales, puesto que el grado de masculinización está determinado por la cantidad de tejido testicular funcional. Estos pacientes tienen un 2,6-4,6% de riesgo de desarrollar una neoplasia gonadal, y más en presencia de un cromosoma Y. Los tumores de células germinales y el disgerminoma constituyen la variedad histológica más frecuente3.
El objetivo es presentar un caso de un paciente masculino de edad media quien comienza por abdomen agudo secundario a masa abdominal en sufrimiento con diagnóstico final de hermafroditismo verdadero más tumor de células germinales en ovario izquierdo.
Caso clínicoPaciente fenotípicamente masculino, de 53 años de edad. Estado civil: unión libre. Profesión: agricultor. Consulta al servicio de urgencias del Hospital Universitario de Caribe por cuadro clínico consistente en dolor abdominal tipo punzada de inicio súbito inicialmente en hemiabdomen izquierdo con posterior generalización asociado a vómitos incontables de contenido alimentario que luego se tornan biliosos, además de presentar aumento progresivo del perímetro abdominal, ausencia de deposiciones y fiebre no cuantificada del mismo tiempo de evolución.
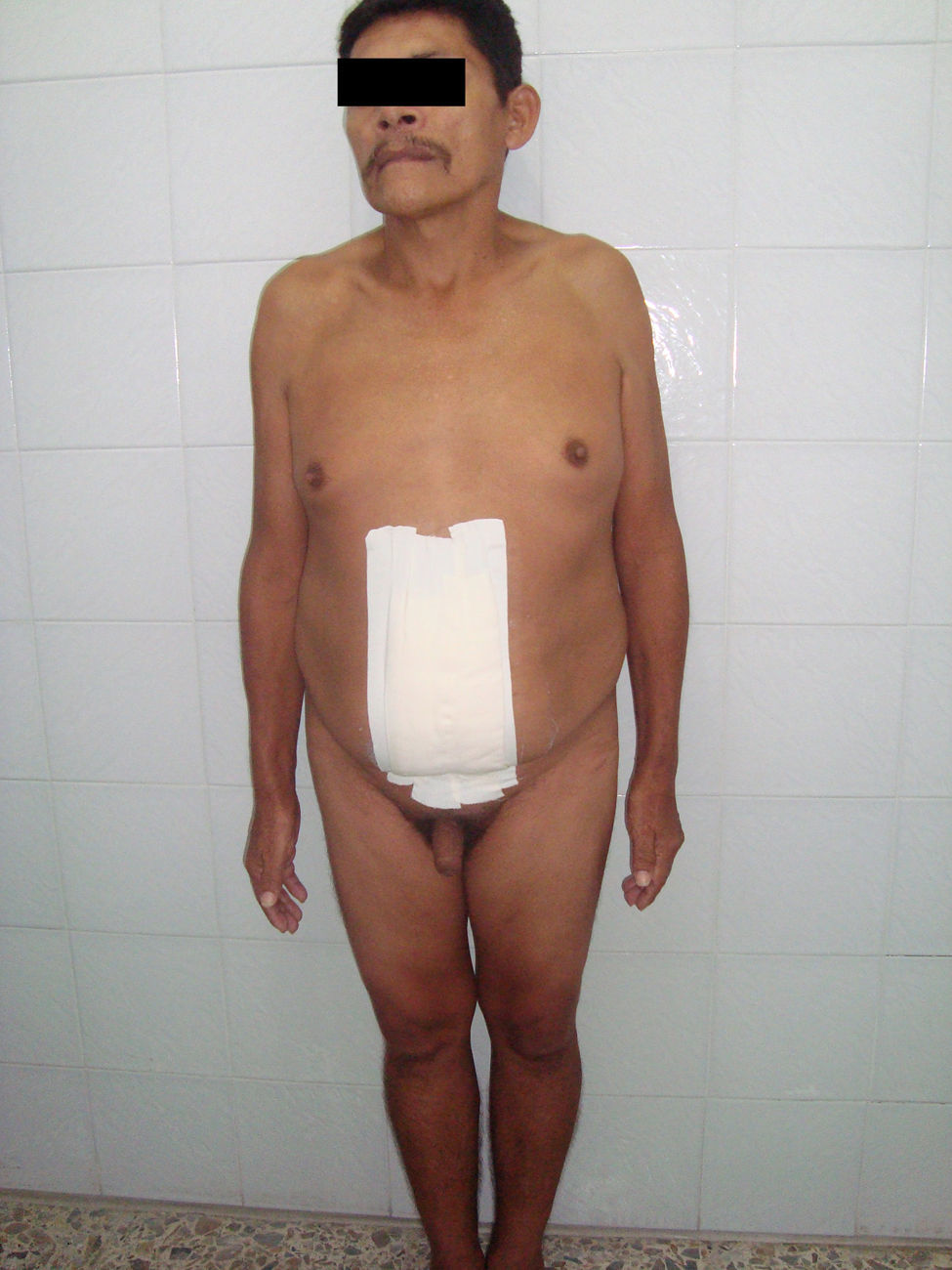
Al examen físico se encuentra paciente en malas condiciones nutricionales, álgido, quejumbroso con gran distensión abdominal, por lo cual se decide tratar con analgésicos sin evidencia de mejoría. Examen genitourinario: Pene de apariencia normal, uretra que desemboca en glande. No se palpan testículos en sacos escrotales (figs. 1 y 2).


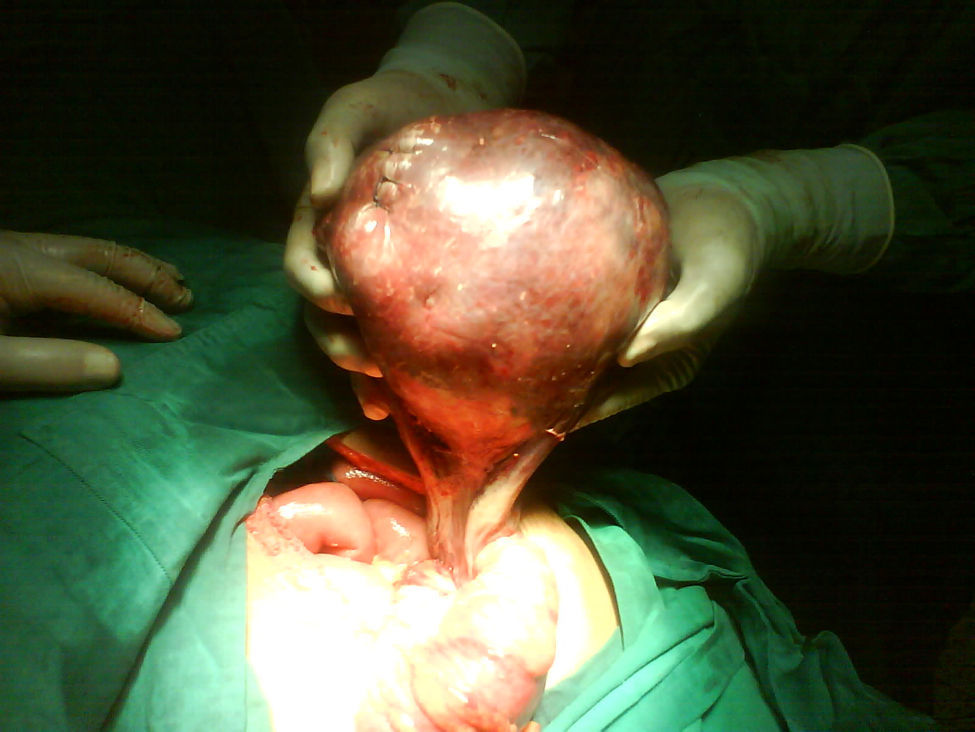
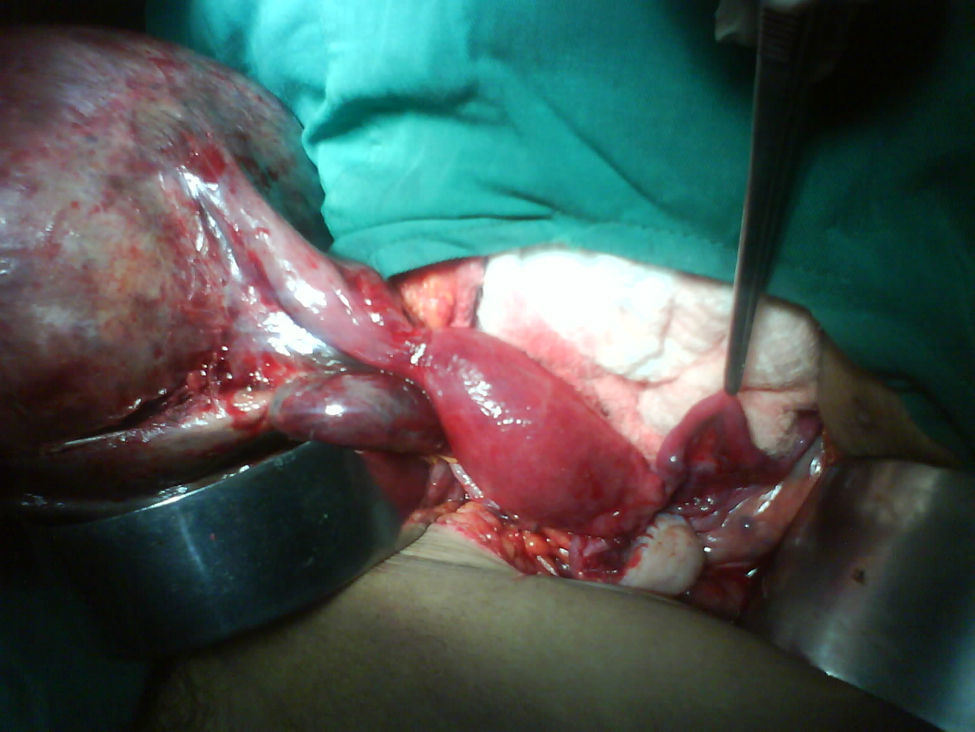
Se decide realizar ecografía, la cual evidencia vesícula dilatada (1.700cc de volumen), próstata aumentada de tamaño (volumen 190cc); entre el hígado y el riñón derecho se observa una masa hipoecogénica sólida que mide 56×37mm y es compatible con neoplasia de glándula suprarrenal derecha, en hipocondrio izquierdo se observa masa heterogénea de 155×45mm compatible con tumor del espacio pararrenal anterior. Adicionalmente se le realiza un tac abdominal simple y contrastado que muestra una gran masa intraabdominal, en flanco izquierdo de 14cm, limitada por una cápsula, con calcificaciones y contenido no homogéneo, con componente líquido mayor y otro graso en su parte superior; en su interior se aprecia septos. La imagen con aspecto de masa tumoral, desarrollada en la topografía de la glándula suprarrenal derecha correspondía a evidente hiperplasia suprarrenal. La glándula suprarrenal izquierda aparece también engrosada. El paciente es sometido a laparotomía exploratoria donde se encuentra masa de aspecto quístico, se bascula la masa y se observa que presenta pedículo torcido correspondiente a trompa de Falopio y ovario izquierdo, se aprecian órganos genitales internos femeninos (ovarios, trompas de Falopio y útero) (figs. 3 y 4).

.")
Estudios de laboratorio complementarios posteriores a los hallazgos quirúrgicos: hormona estimulante del tiroides: 4,26 (RN: 0,270-4,2uUI/l); hormona folículo-estimulante: 20,52 (RN para hombres: 1,5-12,4mUI/l); hormona leutinizante: 19,29 (RN para hombres: 1,7-8,6mUI/l); cortisol sérico matutino, 5:30am: 21,89 (RN: 6,20-16,40), cortisol sérico vespertino 3:00pm: 9,67 (RN: 2,30-11,9ug/dl); prolactina: 8 (hombres: 4,04-15,2; mujeres no embarazadas: 4,79-23,3ng/ml); estradiol: menor de 5 (hombres: 13-54pg/ml); testosterona total: 0,62 (hombres>50: 1,8 a 5,7; mujeres: 0,6-1,1ng/ml); hemoleucograma, ionograma, gases arteriales, función renal y PSA entre límites normales. Cariotipo: 46XX.
Reporte de patología: salpingooforectomía izquierda compatible con teratoma quístico maduro con necrosis hemorrágica y trompa de Falopio con necrosis hemorrágica.
Se decide reintervenir al paciente, sometiéndolo a una histerectomía total cuyo reporte histológico revela lo siguiente: Histerectomía abdominal total con estructuras histológicamente usuales. Cérvix tapizada por epitelio escamoso que conserva su maduración, descansa sobre un estroma fibroso con inflamación crónica leve, acompañada de glándulas endocervicales tapizadas por epitelio cilíndrico sin atipias, endometrio atrófico, ovario y trompa de Falopio histológicamente usual. Citología de secreción intrauterina negativa para malignidad. Salpingooforectomía derecha donde encuentran estructuras histológicamente compatibles con tejido testicular y ovárico (ovoteste).
A pesar de la evidencia de una masa suprarrenal derecha sólida de 55mm, no se tiene reporte de 17-hidroxiprogesterona sérica (17-OHP), lo cual hace evidente la necesidad de investigar en el paciente una hiperplasia suprarrenal congénita. Con todos los datos en mención, se considera que estamos frente a un caso de DDS ovotesticular, antes llamado hermafroditismo verdadero, posiblemente secundario a una hiperplasia suprarrenal congénita, que además nos da evidencias de presentar una de las complicaciones más raras de dicha enfermedad, un teratoma quístico maduro.
DiscusiónEl término hermafroditismo proviene de «Hermafrodito», hijo de Afrodita, diosa del amor y la belleza en la mitología griega, y de Hermes, dios olímpico de las fronteras y los viajeros. Hermafrodito no tenía genitales ambigüos, hasta que una ninfa llamada Salmácide, quien estaba atraída por él, mientras nadaba desnudo, en su contra lo abrazó fuertemente y lo arrastró al fondo suplicando a los dioses que nada los separara. En respuesta a su deseo, ambos cuerpos se fusionaron para siempre en un solo ser, de doble sexo.
El hermafroditismo verdadero constituye una entidad de rara presentación consistente en tejido testicular y ovárico en el mismo paciente. Según la anatomía de las gónadas, el hermafroditismo puede ser: bilateral, cuando las gónadas son ovotestis (es lo más frecuente, cerca del 60%); luego unilateral completo, ovotestis de un lado y ovario del otro; menos frecuentemente se observa un ovotestis de un lado y un testículo del otro; unilateral incompleto, cuando solo existe ovotestis; y alterno cuando existe de un lado un ovario y un testículo del otro4. Se distingue de otras malformaciones genitales por una aberración claramente definida de la cascada endocrina responsable del desarrollo fetal de los genitales externos e internos y por ello tiene potencial de causar alteraciones metabólicas que ponen en riesgo la vida del individuo, cambios físicos inapropiados en la pubertad, confusión de la identidad de género y en algunos casos propensión a sufrir cambios malignos4.
Los pacientes con este tipo de DDS no presentan malformaciones en otros órganos o sistemas, desarrollan inteligencia normal y en general tienen buena expectativa de vida5, aunque, como se mencionó, un pequeño porcentaje (2,6-4,6%) tienen riesgo de desarrollar una neoplasia gonadal, cuya mayor incidencia es en presencia de un cromosoma Y. Los tumores de células germinales son los más frecuentes y el disgerminoma la variedad histológica más frecuente3.
El cariotipo es la herramienta que permite la definición del sexo cromosómico del paciente. Este puede resultar 46XX, el cual es el frecuentemente encontrado (71%), seguido por el mosaicismo 46XX/46XY (20%) y por último 46XY que es infrecuente (7%). En los 2 últimos existen datos de citogenética de cromosoma Y; la presencia de tejido testicular en ellos se explica por el gen SRY, que se considera determinante para la diferenciación testicular, mientras que en el caso de los 46XX el tejido testicular se explica por la presencia oculta del gen SRY debido a la translocación de Y-X o Y-autosoma6,7. En este caso, el sexo cromosómico resultó 46XX, indicando sexo femenino.
El fenotipo del paciente, como se mencionó anteriormente, generalmente es resultado de una alteración en la cascada endocrina que afectó el desarrollo embrionario (desarrollo de conductos de Müller o Wolff) o la producción anormal de 17-OHP en presencia de hiperplasia suprarrenal congénita, principal causa de genitales ambiguos8. A continuación se describen ambas entidades.
La producción de hormona antimülleriana por las células de Sertoli causa regresión de los conductos de Müller. Y la acción de la testosterona y su metabolito dihidrotestosterona producen diferenciación masculina de los conductos de Wolff. Del conducto de Wolff se diferencian: Estructuras masculinas, epidídimo, conducto deferente, vesícula seminal, conducto eyaculatorio y del conducto de Müller. Estructuras femeninas, trompa de Falopio, útero y tercio superior de la vagina. Cuando hay alteración en la producción de alguna de estas hormonas puede haber persistencia de órganos derivados de ambas estructuras. Para detectar las estructuras müllerianas se requiere: ultrasonido y genitograma y en casos muy específicos, RMN.
La hiperplasia suprarrenal congénita es una dolencia que se refiere a cualquiera de las enfermedades autosómicas recesivas que resultan de la mutación genética para enzimas que median los pasos de la producción de cortisol. El 95% es por déficit de la enzima 21-alfa-hidroxilasa. Se presenta en el 75% de los pacientes como nefropatía perdedora de sal, que puede llevar a deshidratación, colapso cardiovascular y muerte. Es la principal causa de genitales ambiguos; un neonato con intersexo tiene un 90% de posibilidad de que este sea secundario a hiperplasia suprarrenal. Esto porque en niñas hay masculinización de los genitales externos por la exposición a una mayor producción de testosterona9. Es posible su diagnóstico prenatal por amniocentesis con niveles elevados de 17-OHP que se encuentra en el camino del metabolismo de colesterol hacia la síntesis de cortisol. Cuando se bloquea la conversión de la hidroxiprogesterona por falla genética de alguna de las enzimas, sus niveles se elevan en sangre. También se puede diagnosticar mediante la medición de la androstenediona o biopsia de vellosidades coriónicas y análisis del ADN, pues se tienen identificados los genes específicos etiológicos.
El tratamiento de los pacientes con hermafroditismo verdadero debe tener un enfoque médico, quirúrgico y psicosocial9. En caso de intersexo con nefropatía perdedora de sal, hay que dar tratamiento médico y solucionar lo que ponga en riesgo la vida. En cuanto al ámbito quirúrgico, se recomienda gonadectomía temprana, sobre todos en los casos de disgenesia gonadal que tengan componente Y; el tiempo de asignación o reasignación quirúrgica de género está en debate. El trastorno de identidad de género que se puede presentar se llama disforia de género, «atrapados en un cuerpo que no es de su sexo», por lo cual es tan importante la intervención en todas las esferas mencionadas10.
ConclusiónSe presenta el caso de un paciente adulto fenotípicamente masculino, al cual se le diagnostica hermafroditismo verdadero, cariotipo 46XX. Hallazgo quirúrgico de estructuras genitales femeninas bilaterales: útero (cervicitis crónica leve, endometrio atrófico), ovarios, trompas de Falopio. Se halla además teratoma quístico maduro con necrosis hemorrágica en gónadas izquierdas. Se descartó hermafroditismo femenino porque en un reporte de cistoscopia no se encontraron los testículos en saco, y posteriormente no se menciona su hallazgo quirúrgico o en las ecografías y tac realizados. Se descartó disgenesia gonadal por no presentar componente Y en cariotipo (46, XX). Además de ello, el paciente tiene evidencia de masa suprarrenal derecha sólida de 55mm, pero no tiene reporte de 17-OHP por lo cual sería conveniente investigar en él si la masa sería la causa de hiperplasia suprarrenal como agente de la virilización.
Además, el paciente presenta una de las complicaciones más raras de dicho desarreglo, un teratoma quístico maduro, tumor germinal que ocurre del 2,6-4,6% de los casos según reporta la literatura.
Se le hizo tratamiento quirúrgico, con gonadectomía y resección tumoral, con buena evolución y pronóstico. Además se le ordenó intervención psicológica: intervención cognitiva-conductual-restructuración cognitiva; intervención humanista-técnica sistémica familiar e intervención cognitiva conductual-desensibilización sistemática, para tratar integralmente el cuadro.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónRecursos propios.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.