



El síndrome de Peutz-Jeghers (PJ) es un desorden raro, autosómico dominante, caracterizado por la presencia de múltiples pólipos hamartomatosos, pigmentación mucocutánea y un incremento en la susceptibilidad para desarrollar diferentes tumores. Existe una asociación poco frecuente de esta con el desarrollo de tumores testiculares de células de Sertoli que usualmente son bilaterales y multifocales y constituyen un elemento de alta importancia durante la valoración de pacientes con esta enfermedad. Se reportan 2 casos de presentación rara e inusual, uno de ellos sin presentación polipósica de la enfermedad y otro con desarrollo de enfermedad hipofisiaria, así como su manejo, correlacionándolo con lo reportado en la literatura hasta la fecha.
Pacientes con PJ tienen un mayor riesgo de desarrollar tumores en diferentes órganos y, aunque los testículos son un sitio poco común, su incidencia es mucho más alta que en la población general, por lo tanto se debe realizar monitoreo en este grupo de pacientes con el autoexamen y la visita regular para buscar activamente los signos que pueden indicar el desarrollo de la enfermedad testicular. El tipo histológico más común es el de células de Sertoli que, por lo general, tiene un comportamiento benigno. Hasta el momento no hay marcadores histológicos, biológicos ni de imagen que permitan predecir su comportamiento ni su potencial metastásico. Su tratamiento quirúrgico ha cambiado: la orquiectomía parcial, y no la orquiectomía radical, se propone como una opción viable cuando el parénquima testicular restante es adecuado para su conservación.
Peutz-Jeghers (PJ) syndrome is an autosomal dominant disorder characterised by the presence of multiple hamartomatous polyps, mucocutaneous pigmentation, and an increased susceptibility to develop different tumours. It is associated with the development of testicular Sertoli cell tumours, which are usually bilateral and multifocal and are a highly important element in the evaluation of patients with this disease. Two cases of rare and unusual presentation are presented; one of them without the polyp presentation of the disease and the other one with pituitary disease, along with their management, and a correlation with the information reported in the literature to date.
PJ patients have an increased risk of developing tumours in different organs, and although testicles are an uncommon site, its incidence is much higher than that of the general population, therefore monitoring should be performed in this group of patients with self-examination and regular visits, in order to actively look for signs that may indicate development of testicular disease. The most common histological type is the Sertoli cells tumour, which usually has a benign behaviour. So far, there are no histological, biological or imaging markers that predict their behaviour or metastatic potential. Their surgical management has changed according to the most recent literature reviewed, with partial orchiectomy and not radical orchiectomy being proposed as a viable option when the remaining testicular parenchyma is suitable for preservation.
El síndrome de Peutz-Jeghers es un trastorno autosómico dominante caracterizado por múltiples pólipos hamartomatosos gastrointestinales, pigmentación mucocutánea y una mayor predisposición a varias neoplasias. Existe una asociación de esta entidad con tumores calcificantes de células de Sertoli (TCCG), usualmente de presentación bilateral y multifocal. Los pacientes con PJ tienen un riesgo aumentado de desarrollar tumores en diferentes órganos, siendo los testículos un sitio infrecuente pero con una incidencia mucho mayor que en la población general. Los pacientes con PJ tienen un riesgo relativo global de desarrollar cáncer 15 veces mayor que el de la población general1.
Los tumores de células de Sertoli pueden presentarse de manera espontánea en un 60% de los casos, pero existe una asociación con síndromes genéticos claramente conocida: el síndrome de PJ y el complejo de Carney son los más referenciados en la literatura2.
Las manifestaciónes clínicas de estos tumores incluyen ginecomastia prepuberal y calcificaciones testiculares, principalmente3. Los tumores testiculares originados de los cordones sexuales son extremadamente raros y constituyen solo el 5% de todas las neoplasias del testículo. Las alteraciones hormonales son secundarias al aumento en la producción de estrógenos y de expresión de aromatasa. Se ha propuesto a la inhibina alfa como marcador tumoral.
El manejo de los TCCG y su evaluación de potencial de malignidad es de gran importancia. El tratamiento se ha modificado en los últimos años: de un manejo quirúrgico con principios oncológicos a un manejo médico conservador que se dirige a reducir los efectos estrogénicos sobre el tejido mamario y el crecimiento óseo. Los inhibidores de la aromatasa son la mejor opción para lograr esta meta3–5.
ObjetivoPresentar 2 casos de pacientes con síndrome de PJ, quienes desarrollaron tumores testiculares, y el manejo institucional, correlacionándolo con lo descrito en la literatura en la actualidad.
Materiales y métodosSe realizó una revisión de la literatura que incluyera los términos «Peutz-Jeghers, Sertoli Tumors» desde enero del 2004 hasta enero del 2014 en la base de datos de PubMed, en los idiomas inglés y español, encontrando un total de 25 publicaciones. Se procedió a identificar las publicaciones de relevancia para la revisión: un total de 10 publicaciones fueron revisadas y referenciadas. Adicionalmente, se presentan 2 casos de pacientes que fueron atendidos en nuestras instituciones los cuales se comparan, analizan y describen a partir de los hallazgos e intervenciones reportados hasta la fecha en la literatura.
Caso clínico 1Paciente de 9 años y 3 meses de edad con antecedente de enfermedad de PJ diagnosticada a los 5 años de edad, hasta el momento sin manifestaciones intestinales, confirmado por un estudio colonoscópico reciente normal (hallazgo infrecuente en el síndrome de PJ). Consulta por cuadro de 2 años de evolución de cambios en la consistencia de ambos testículos, con asimetría asociada y desarrollo puberal acelerado.
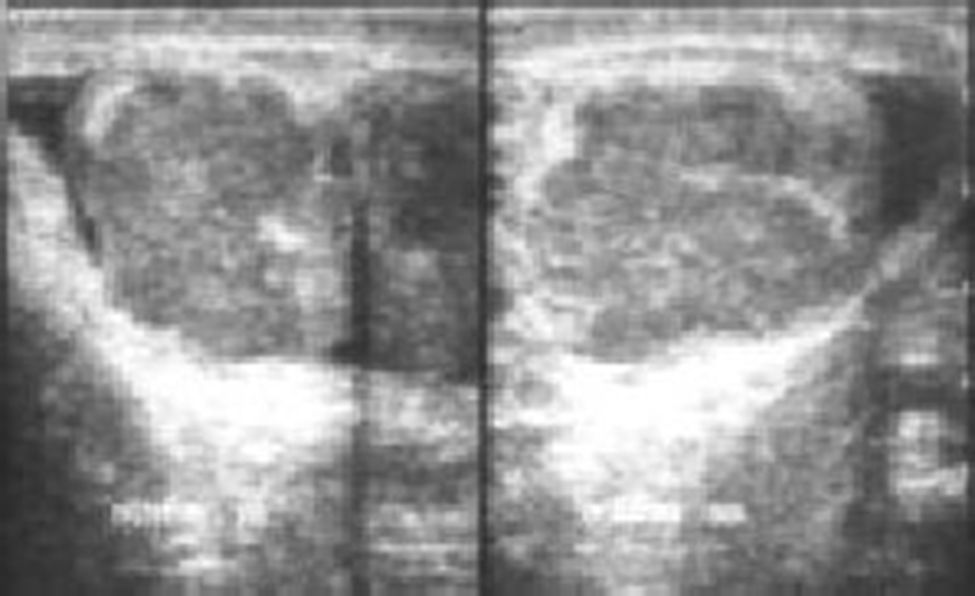
Presentaba lesiones lentiginosas en cara, labios, pies y ano, ginecomastia bilateral, estadio puberal A3P4 Tanner 3 con gónadas palpables irregulares a la palpación de 20ml de volumen bilateral. Estatura de 1,47m, talla para la edad <2 desviaciones estándar; en la ecografía testicular bilateral se reportó el testículo derecho con un tamaño de 48×36×24mm y un volumen de 23 cc, y el testículo izquierdo con un tamaño de 24×29×32mm y un volumen de 35 cc. Ambos testículos presentaban múltiples masas bilaterales de hasta 20mm de diámetro mayor, sin lograr diferenciar el parénquima testicular de forma adecuada (fig. 1). En la tomografía de abdomen y pelvis contrastada no se aprecian lesiones retroperitoneales y fue reportada como normal.

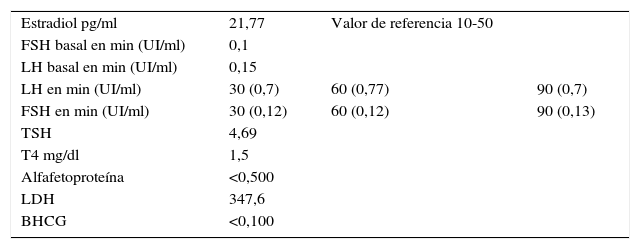
El perfil hormonal muestra niveles de estradiol similares a los reportados en la literatura dentro de la normalidad (tabla 1).
Perfil hormonal del paciente 1 al momento del diagnóstico
| Estradiol pg/ml | 21,77 | Valor de referencia 10-50 | |
| FSH basal en min (UI/ml) | 0,1 | ||
| LH basal en min (UI/ml) | 0,15 | ||
| LH en min (UI/ml) | 30 (0,7) | 60 (0,77) | 90 (0,7) |
| FSH en min (UI/ml) | 30 (0,12) | 60 (0,12) | 90 (0,13) |
| TSH | 4,69 | ||
| T4 mg/dl | 1,5 | ||
| Alfafetoproteína | <0,500 | ||
| LDH | 347,6 | ||
| BHCG | <0,100 |
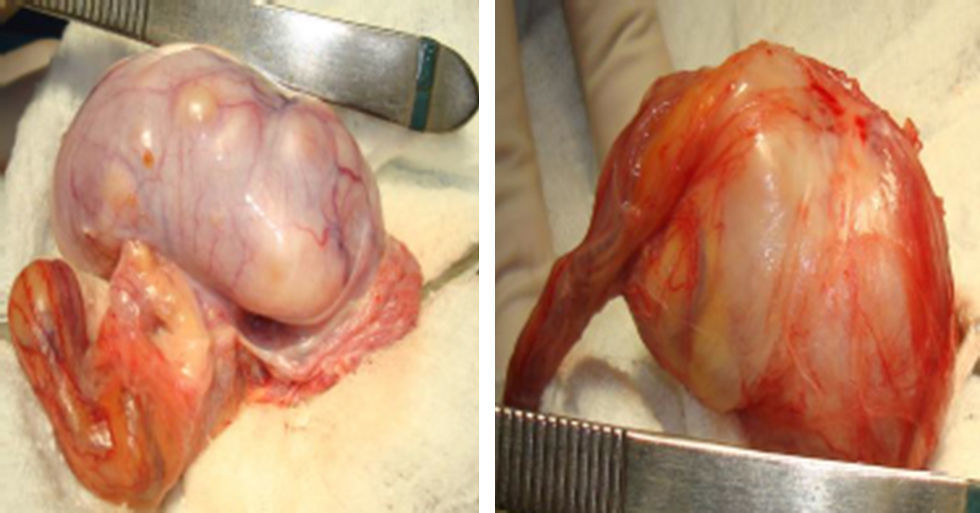
El paciente es llevado a biopsia testicular por congelación a cielo abierto en la que se aprecian unos testículos aumentados de tamaño de contornos irregulares con parénquima remplazado por múltiples lesiones nodulares sólidas y quísticas (figs. 2 y 3). Se obtiene como reporte un tumor del estroma gonadal que indica tumor de células de Sertoli, por lo que se decide practicar una orquiectomía radical bilateral, dado el completo remplazo del parénquima testicular.

Tumor de células de Sertoli, variante grande, calcificante, con compromiso del 95% del parénquima testicular en contacto con la túnica albugínea sin sobrepasarla, sin evidencia de invasión linfovascular ni perineural. Mitosis 1 por campo, sin atipia celular marcada (fig. 4).
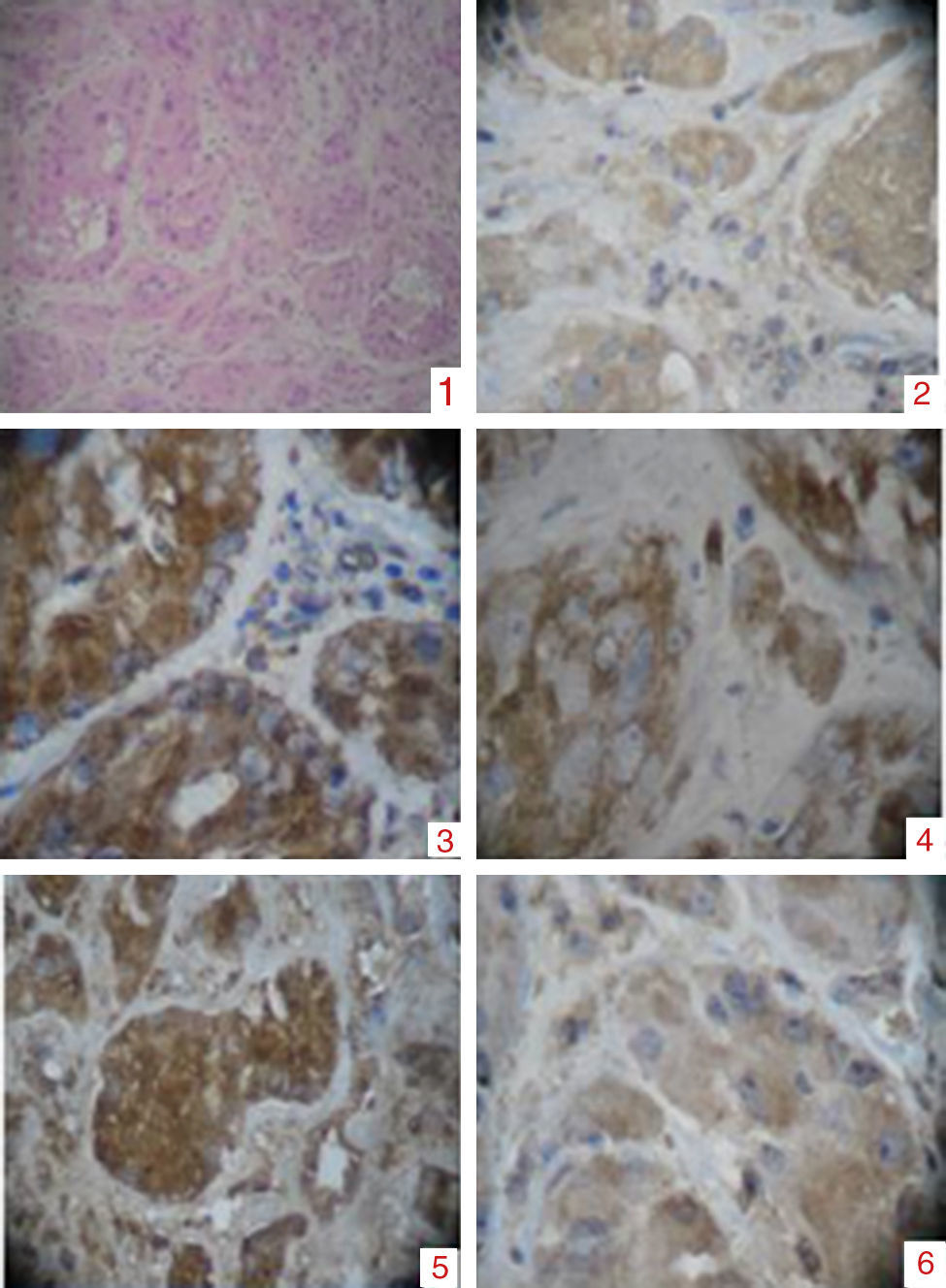
 Túbulos seminíferos dilatados, constituidos por células poligonales intensamente eosinofílicas, los núcleos se reconocen redondos con nucléolos prominentes. 2) Tinción con alfafetoproteína. 3) Tinción con vimentina. 4) Tinción con inhibina. 5) Tinción con BHCG. 6) Bajo índice de proliferacion mitótica.")
Placa de hematoxilina: se observa pérdida de la arquitectura. 1) Túbulos seminíferos dilatados, constituidos por células poligonales intensamente eosinofílicas, los núcleos se reconocen redondos con nucléolos prominentes. 2) Tinción con alfafetoproteína. 3) Tinción con vimentina. 4) Tinción con inhibina. 5) Tinción con BHCG. 6) Bajo índice de proliferacion mitótica.
El paciente inicia suplencia hormonal a la edad de 12 años, luego de llevar un año de seguimiento y de asegurar una maduración esquelética completa con enanato de testosterona. Durante el seguimiento no se registraron lesiones metastásicas ni compromiso a distancia. No requirió bloqueo estrogénico adicional, ya que los niveles iniciales de estrógenos eran normales.
Caso clínico 2Paciente con antecedente de síndrome de PJ con manifestación polipomatosa intestinal clásica quien, a la edad de 14 años, refiere aumento del tamaño de ambos testículos de 3 meses de evolución, con hallazgo en ecografía testicular de alteración en la ecogenicidad debido a masas ecogénicas multifocales bilaterales con múltiples calcificaciones de pequeño tamaño, que se distribuyen difusamente. Adicionalmente se le diagnostica un adenoma hipofisario que requiere extirpación quirúrgica en 2 oportunidades por recidiva.
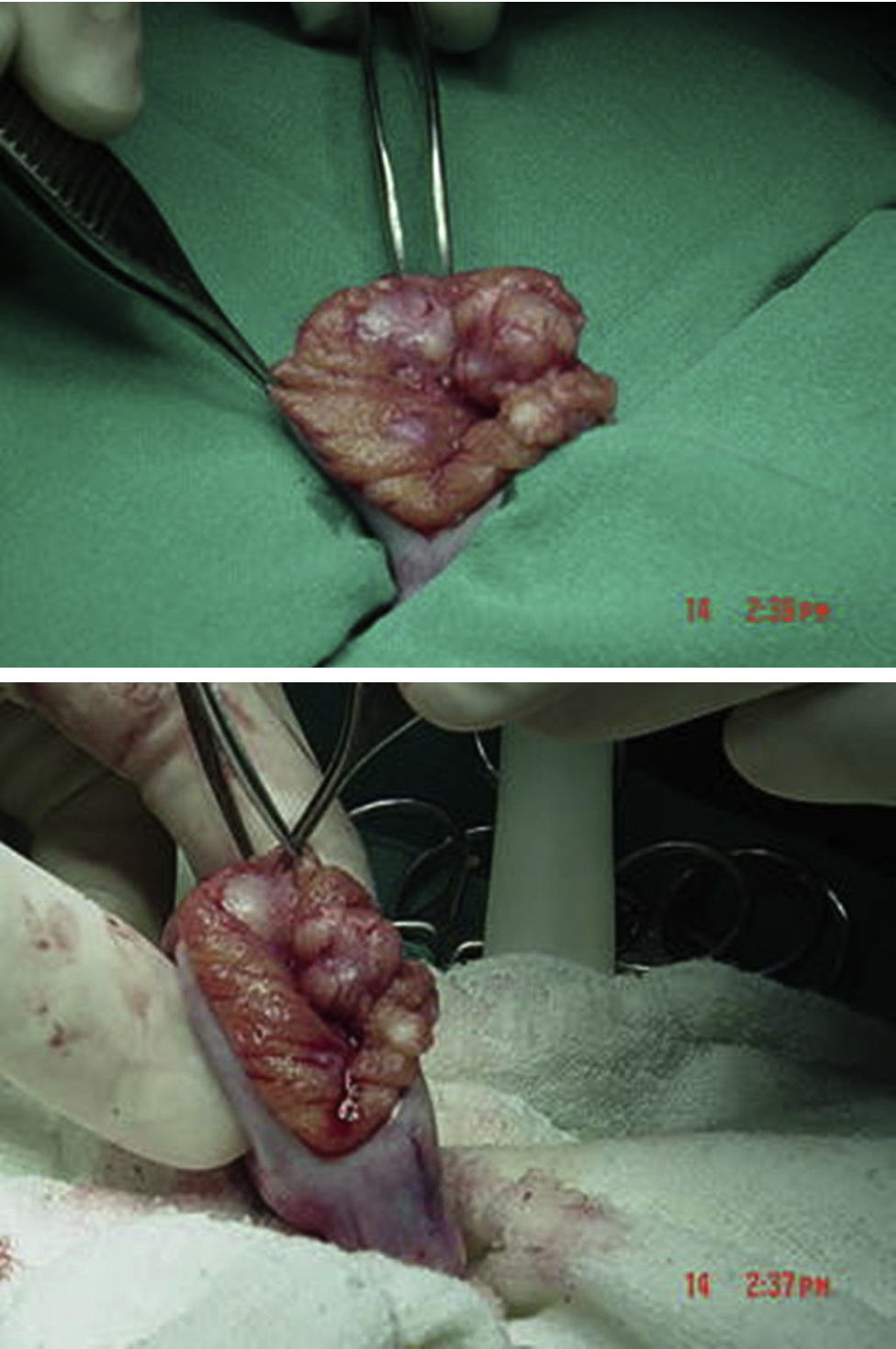
Se encuentra perfil hormonal con testosterona en rango puberal con estradiol normal (tabla 2). Resto de marcadores negativos. Se decide entonces realizar biopsia inguinal testicular bilateral, a cielo abierto por congelación por vía inguinal en junio del 2004, encontrando testículos con compromiso tumoral bilateral generalizado con reporte de biopsia por congelación compatible con tumor de células de Sertoli (figs. 5 y 6). Por estos hallazgos se decide realizar orquiectomía radical bilateral.

Tumor de células de Sertoli variante grande calcificante con compromiso del 80% del parénquima, sin evidencia de invasión linfovascular ni perineural. Mitosis 3 por campo sin atipia celular marcada.
El paciente completa ya 9 años de seguimiento sin documentarse recaída tumoral ni compromiso metastásico. Se encuentra en suplencia hormonal permanente con enanato de testosterona por parte de endocrinología.
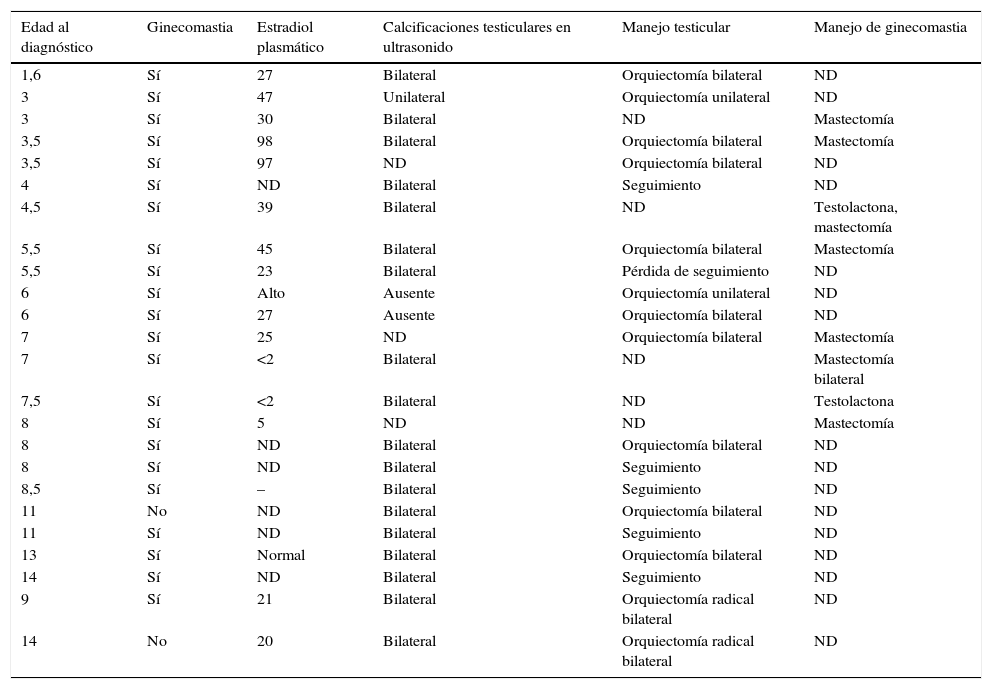
DiscusiónEl síndrome de PJ es un desorden autosómico dominante, clásicamente asociado a una poliposis gastrointestinal, pigmentación cutánea y predisposición aumentada a presentar enfermedad tumoral. El gen humano LKB1/STK11 codifica una serina/treonina protein-cinasa que está defectuosa en los pacientes con este síndrome6. Diferentes revisiones de la literatura (tabla 3) han identificado más de 40 mutaciones del LKB1 en tumores somáticos y 41 mutaciones en tumores esporádicos y de línea germinal, lo que explicaría la predisposición aumentada en este grupo de pacientes a manifestaciones tumorales extragastrointestinales6,7.
Recopilación de reportes de casos de la literatura hasta enero de 2014. Se muestra intervención, hallazgos encontrados y manejo endocrinológico, de haber sido mostrado. Los 2 casos presentados se recogen en las casillas 23 y 24
| Edad al diagnóstico | Ginecomastia | Estradiol plasmático | Calcificaciones testiculares en ultrasonido | Manejo testicular | Manejo de ginecomastia |
|---|---|---|---|---|---|
| 1,6 | Sí | 27 | Bilateral | Orquiectomía bilateral | ND |
| 3 | Sí | 47 | Unilateral | Orquiectomía unilateral | ND |
| 3 | Sí | 30 | Bilateral | ND | Mastectomía |
| 3,5 | Sí | 98 | Bilateral | Orquiectomía bilateral | Mastectomía |
| 3,5 | Sí | 97 | ND | Orquiectomía bilateral | ND |
| 4 | Sí | ND | Bilateral | Seguimiento | ND |
| 4,5 | Sí | 39 | Bilateral | ND | Testolactona, mastectomía |
| 5,5 | Sí | 45 | Bilateral | Orquiectomía bilateral | Mastectomía |
| 5,5 | Sí | 23 | Bilateral | Pérdida de seguimiento | ND |
| 6 | Sí | Alto | Ausente | Orquiectomía unilateral | ND |
| 6 | Sí | 27 | Ausente | Orquiectomía bilateral | ND |
| 7 | Sí | 25 | ND | Orquiectomía bilateral | Mastectomía |
| 7 | Sí | <2 | Bilateral | ND | Mastectomía bilateral |
| 7,5 | Sí | <2 | Bilateral | ND | Testolactona |
| 8 | Sí | 5 | ND | ND | Mastectomía |
| 8 | Sí | ND | Bilateral | Orquiectomía bilateral | ND |
| 8 | Sí | ND | Bilateral | Seguimiento | ND |
| 8,5 | Sí | – | Bilateral | Seguimiento | ND |
| 11 | No | ND | Bilateral | Orquiectomía bilateral | ND |
| 11 | Sí | ND | Bilateral | Seguimiento | ND |
| 13 | Sí | Normal | Bilateral | Orquiectomía bilateral | ND |
| 14 | Sí | ND | Bilateral | Seguimiento | ND |
| 9 | Sí | 21 | Bilateral | Orquiectomía radical bilateral | ND |
| 14 | No | 20 | Bilateral | Orquiectomía radical bilateral | ND |
Los pólipos son generalmente hamartomas benignos localizados en cualquier lugar del intestino delgado o grueso. El 88% de los pacientes con PJ tienen pólipos intestinales, siendo poco usual la presentación sin estas lesiones1,8-11. La hiperpigmentación es común en la mucosa perioral y perianal, labios, dorso de la mano, dedos, palmas y pies. Este signo está presente desde el nacimiento o aparece durante la infancia y su intensidad puede disminuir con el tiempo. Los pacientes con PJ tienen un riesgo relativo global de desarrollar cáncer 15 veces mayor que el de la población general1,12,13. Dentro de estos se encuentra el cáncer de intestino, colon, páncreas, estómago, pulmón, seno, cérvix, ovario y testicular, siendo este último muy raro.
La incidencia del PJ es igual para ambos sexos. La presentación de pacientes con PJ y tumores testiculares generalmente ocurre entre el nacimiento y los 6 años de edad14. Los tumores de células de Sertoli se manifiestan con ginecomastia y pubertad precoz hasta en el 42% de los pacientes, lo cual se explica por la producción de hormonas esteroideas tanto por las células de Sertoli neoplásicas como por las células de Leydig extratumorales15–17. Otras anormalidades incluyen un aumento en el índice de actividad de aromatasa (estradiol/testosterona), ya que pequeñas dosis de estrógenos pueden estimular el crecimiento óseo. Estos pacientes típicamente tienen una edad y crecimiento óseo avanzado, ya que la estimulación estrogénica persistente lleva a un cierre prematuro de las epífisis de crecimiento, y baja estatura en la edad adulta.
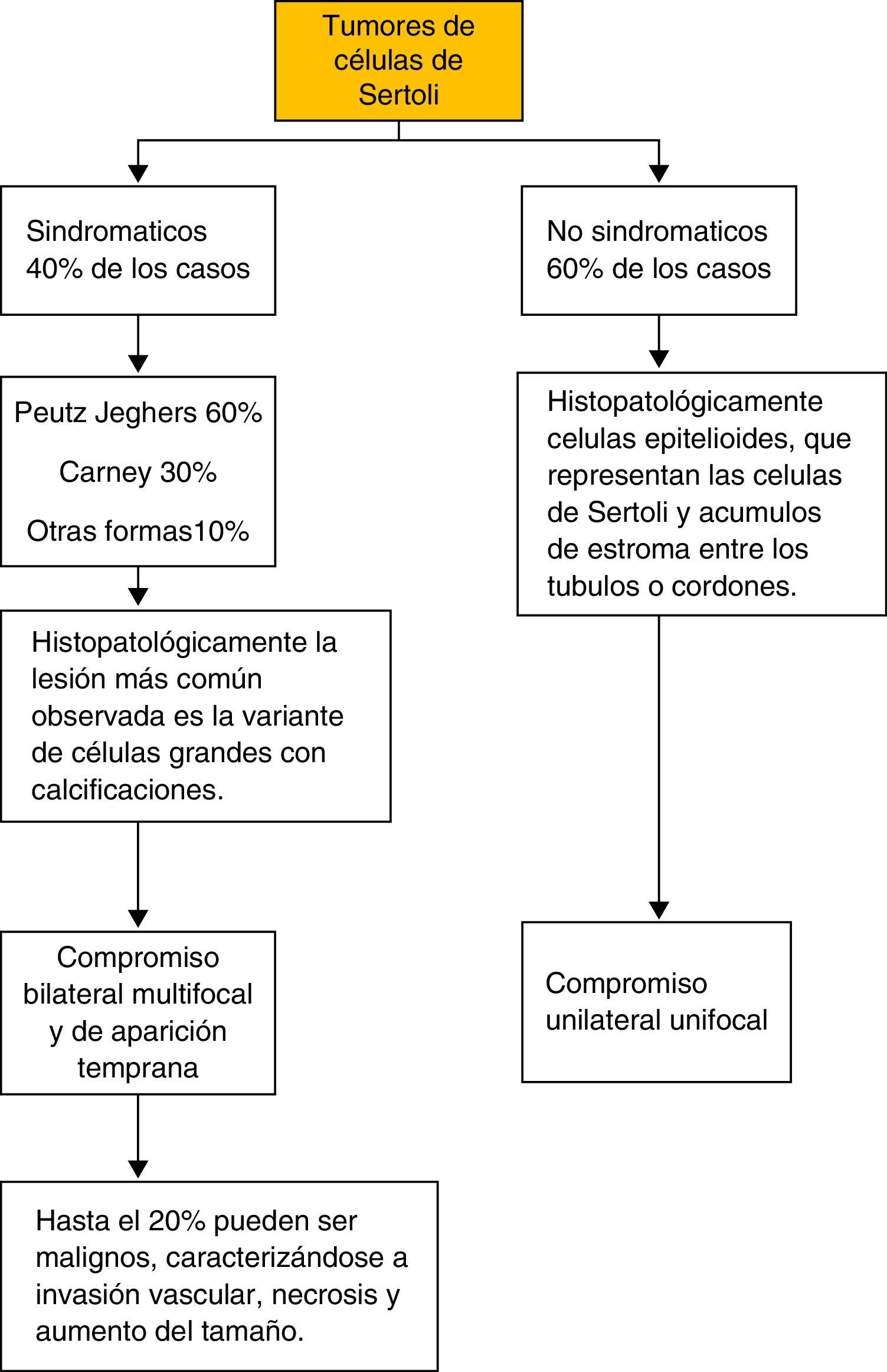
En los tumores de células de Sertoli se reconocen 2 grupos de acuerdo con su forma de presentación (fig. 7). Las formas sindromáticas con carácter hereditario que constituyen el 40% de los casos, en donde el síndrome de PJ constituye un 60% de los casos, y el síndrome de Carney (un 30%). Esta forma de presentación usualmente es bilateral, multifocal y de aparición temprana. Y un segundo grupo, el esporádico, que es el grupo mayoritario, con aproximadamente el 60% de todos los casos. En este grupo, característicamente, la presentación es unilateral y unifocal con lesiones pequeñas asociadas6,18,19. En los tumores de células de Sertoli sindromáticos se debe descartar la presencia de enfermedades asociadas como adenomas hipofisiarios, shwanomas melanóticos, mixomas auriculares, los cuales pueden presentarse simultáneamente en el PJ12.

Los TCCG manifiestan un bajo potencial de malignidad y es poco probable la presentación con lesiones metastásicas1. La capacidad predictiva de comportamiento maligno de los TCCG, asociados o no a síndrome de PJ, basada en hallazgos clínicos, imagenológicos, histopatológicos y de laboratorio, no es buena, lo que ha dificultado predecir claramente cuáles de estos tumores pueden manifestarse con un comportamiento agresivo, pero, en términos generales, su comportamiento es benigno. Se ha descrito en la literatura que aproximadamente el 17% de estos tumores pueden presentar un comportamiento agresivo, principalmente cuando las lesiones son únicas o miden más de 4cm en las presentaciones no sindromáticas2. Otros hallazgos que han sido descritos de mal pronóstico son la extensión tumoral extratesticular, la atipia celular marcada y la mitosis mayor a 3 por campo, pues estos indican un curso más agresivo y requieren un manejo más radical17. Estas observaciones han permitido considerar el manejo conservador con orquiectomía parcial en vez del manejo tradicional con orquiectomía radical en este grupo de tumores y con características claras asociadas12,20.
En los 2 casos reportados se realizó manejo con orquiectomía radical, dado el compromiso multifocal bilateral, el gran tamaño de las lesiones y el parénquima testicular residual escaso que no permitía la realización de procedimientos conservadores.
El diagnóstico imagenológico se realiza a través de ultrasonido, evidenciando masas difusas, heterogéneas con calcificaciones difusas. Debe ponerse énfasis en la evaluación del parénquima testicular residual para basar decisiones terapéuticas.
La terapia médica en este grupo de pacientes se fundamenta en reducir los efectos del aumento de los estrógenos sobre la glándula mamaria y el sistema óseo, siendo los inhibidores de la aromatasa la mejor opción para alcanzar este objetivo3,19,21. El anatrozol, un inhibidor de la aromatasa, es uno de los medicamentos que ha demostrado un buen perfil de seguridad y efectividad en el control de las manifestaciones clínicas de los tumores de células de Sertoli asociado a síndrome de PJ3. Existe la posibilidad de manejo con orquiectomía para tratar la progresión rápida de la maduración esquelética en casos refractarios basados en el potencial de malignidad de esta enfermedad y la pobre respuesta al manejo médico. Se desconoce el potencial de desarrollar cáncer de seno en pacientes con ginecomastia, síndrome de PJ y tumores testiculares. La terapia médica con antiestrógenos en monoterapia no es efectiva para tratar la ginecomastia prepuberal, ante un cuadro avanzado el tratamiento debe incluir una mastectomía. La radioterapia no es recomendada3.
ConclusiónLos pacientes con PJ tienen un riesgo aumentado de desarrollar tumores en diferentes órganos, siendo los testículos un sitio infrecuente pero con una incidencia mucho mayor que en la población general. Por esta razón debe realizarse seguimiento en este grupo de pacientes con autoexamen y visita periódica en busca de signos que indiquen desarrollo de enfermedad testicular. Hasta el momento no se cuenta con marcadores histológicos, biológicos ni imagenológicos que predigan su comportamiento o potencial metastásico. Su manejo quirúrgico se ha modificado según lo encontrado en las publicaciones más frecuentes y se expone la orquiectomía parcial, y no la radical, como una opción viable cuando el parénquima testicular restante sea susceptible de preservación. Ninguno de los casos presentados fue susceptible de manejo preservador de parénquima testicular por su gran tamaño, ni de bloqueo estrogénico, ya que los niveles de estradiol eran normales o ligeramente elevados al momento del diagnóstico. Todos los pacientes con síndrome de PJ deben recibir chequeo periódico de los testículos en búsqueda de anormalidades a la palpación. Los pacientes con tumores menores de 4cm, índice mitótico inferior a 3 por campo, baja atipia, sin evidencia de compromiso extratesticular y con compromiso bilateral y parénquima testicular residual imagenológicamente no comprometido pueden ser tratados mediante procedimientos conservadores como la orquiectomía parcial.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.