


Algunos genes relacionados con proteínas que participan en algunas rutas metabólicas podrían tener un papel en el desarrollo de los síndromes coronarios agudos.
ObjetivoCorrelación entre polimorfismos y su relación con eventos adversos en síndromes coronarios agudos.
MétodoProspectivo, seguimiento hospitalario y a un año. Inclusión: síndromes coronarios agudos con desnivel o elevación del ST secundario a aterotrombosis, estabilidad clínica. En todos, reacción de cadena de polimerasa y polimorfismos de la longitud de los fragmentos de restricción. Al estandarizar reacciones de cadena y genotipificación se realizó análisis preliminar de distribución de genotipos para cada polimorfismo y en ninguno se observaron desviaciones en la ley de equilibrio de Hardy-Weinberg (p>0,05).
ResultadosDe 2003 a 2005 se ingresaron 150 sujetos. Se analizaron 14 polimorfismos en 9 genes (fibrinógeno, factor v, vii, ii, xiii, activador e inhibidor del plasminógeno-1 y proteína C reactiva). En síndromes coronarios agudos, un fibrinógeno>450mg/dL y leucocitos>8, 500 cél/mm3 fueron marcadores de mal pronóstico a un año. Los análisis de regresión identificaron al −148 CT/TT del fibrinógeno y al −717 AG/GG de la proteína C reactiva como marcadores de isquemia recurrente y al 1691GA+AA para reinfarto.
ConclusiónEn pacientes con síndromes coronarios agudos se demostró una relación entre polimorfismos relacionados con hemostasia e inflamación con eventos adversos, por lo que podrían considerarse marcadores de enfermedad coronaria. Se requiere una muestra mayor para confirmar estos resultados.
The genes coding for proteins due to their activity in several metabolic pathways could be related with the onset of acute coronary syndromes.
ObjectiveRelationship among polymorphisms and adverse events in.
MethodsProspective. In – hospital, one – year follow-up. Inclusion Acute coronary syndromes with ST elevation or depression secondary to atherothrombosis, clinical stability. In all, polymerase chain reaction and length polymorphism of restriction fragments. By standardizing chain reactions and genotyping,a preliminary analysis of distribution of genotypes was performed for each polymorphism and no deviations were observed in the law of Hardy-Weinberg equilibrium (P>.05).
ResultsFrom 2003 to 2005, 150 subjects were enrolled. We analyzed 14 polymorphisms in 9 genes (fibrinogen, factor v, vii, ii, xiii, plasminogen activator and inhibitor-1, C-reactive protein). In acute coronary syndromes, fibrinogen>450mg/dL and white blood count 8500 cells/mm3 were markers of poor prognosis to one year. Regression analysis identified the −148 CT/TT and fibrinogen −717 AG/GG of C-reactive protein as a marker of recurrent ischemia and reinfarction 1691GA + AA.
ConclusionWe are showing a relationship among polymorphisms involved in inflammation and hemostasis with adverse events in the acute phase and follow-up in acute coronary syndromes patients that could be considered as markers of ischemic heart disease. Larger sample is needed to confirm these results.
Los síndromes coronarios agudos (SCA) son un término que da identidad a una constelación de síntomas secundarios a isquemia inducida por trombosis aguda (>85%)1,2. Su espectro fisiopatogénico (aterotrombosis) incluye una compleja interacción entre medio ambiente, inflamación, disfunción endotelial y mensajes moleculares. En un extremo del espectro, la trombosis depende de una interacción entre factores de inflamación y hemostáticos que pueden contribuir a la extensión y persistencia del trombo, al fracaso terapéutico y a eventos cardiovasculares adversos mayores (ECAM)3. En el otro extremo, los polimorfismos por su actividad en algunas rutas metabólicas se relacionan con el inicio de la enfermedad y por su participación en inflamación, trombosis y trombo-resistencia podrían tener importantes implicaciones en la evolución. Por esta evidencia y por su importancia en prevención primaria y secundaria realizamos un estudio prospectivo para determinar la relación entre polimorfismos y ECAM.
MétodosEstudio prospectivo de cohortes con seguimiento hospitalario y a un año. Objetivo principal: correlación entre polimorfismos (marcadores de enfermedad coronaria [EC]) inestable y estable) y su relación con ECAM en SCA. Objetivos secundarios: a) frecuencia en 3 cohortes, SCA, EC estable (ECE) y sujetos normales; b) identificar en fase aguda parámetros bioquímicos y hematológicos y compararlos con controles; c) determinar y correlacionar polimorfismos con los niveles plasmáticos de factores bioquímicos y hematológicos, y ECAM; y d) seguimiento en el grupo de SCA para ECAM adversos. Inclusión: a) edad entre 35 y 75 años; b) SCA con desnivel o elevación del ST, con o sin necrosis secundario a aterotrombosis demostrado angiográficamente; c) estabilidad clínica. Exclusión: a) >75 años; b) SCA secundario a estrés, anemia, etcétera; c) Killip clase >ii; d) SCA en los 3 últimos meses; e) fracción de expulsión<35%; f) enfermedad hematológica, hepática o neoplásica; g) actividad inflamatoria aguda o crónica; y h) consumo excesivo de alcohol.
ControlesEnfermedad coronaria estable: pacientes con diagnóstico previo de cardiopatía isquémica por aterosclerosis coronaria demostrada angiográficamente, sin dolor con perfil isquémico en reposo o ejercicio y sin expresión electrocardiográfica de isquemia aguda en los últimos 6 meses. Sujetos sanos: sin enfermedad cardiovascular excluida por historia clínica, examen físico, signos vitales, laboratorio, radiografía de tórax, electrocardiograma y ecocardiograma. Ambos grupos se eligieron por edad y sexo con relación al grupo en estudio.
El protocolo y el consentimiento informado fueron aprobados por los Comités de Investigación y Ética del Hospital de Cardiología No 34 y Hospital Universitario Dr. José E. González de la Universidad Autónoma de Nuevo León. Todos los pacientes leyeron y firmaron el consentimiento informado.
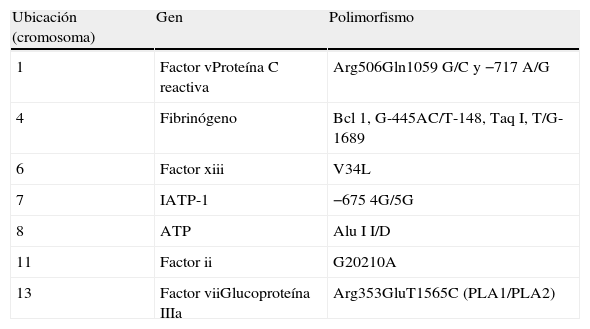
Selección de genesPor la complejidad de los polimorfismos la selección genética (rutas metabólicas) se realizó utilizando datos previamente publicados en donde demostramos asociación entre marcadores bioquímicos (fibrinógeno, plasminógeno, antitrombina iii, proteína C activada, proteína S, inhibidor de la proteína C, a2-antiplasmina y proteína C reactiva) con ECAM3,4 (tabla 1).
Genes y polimorfismos estudiados
| Ubicación (cromosoma) | Gen | Polimorfismo |
| 1 | Factor vProteína C reactiva | Arg506Gln1059 G/C y −717 A/G |
| 4 | Fibrinógeno | Bcl 1, G-445AC/T-148, Taq I, T/G-1689 |
| 6 | Factor xiii | V34L |
| 7 | IATP-1 | −675 4G/5G |
| 8 | ATP | Alu I I/D |
| 11 | Factor ii | G20210A |
| 13 | Factor viiGlucoproteína IIIa | Arg353GluT1565C (PLA1/PLA2) |
ATP: activador tisular del plasminógeno; IATP-1: inhibidor del activador tisular del plasminógeno 1.
Se extrajo mediante la técnica TSNT (Tritón X-100. SDS. NaCl y Tris-EDTA) previamente descrita5.
Reacciones de cadena de polimerasa de los genesCiclos con etapas de desnaturalización, alineamiento y extensión. En presencia de la TaqDNA polimerasa, MgCl2 y dNTPs, los oligonucleótidos se tornan híbridos con la secuencia blanco específica5. En la detección de los genotipos se utilizaron oligonucleótidos específicos.
Polimorfismos de la longitud de los fragmentos de restricciónSe detectaron fragmentos de ADN posterior a su digestión por enzimas de restricción. Los productos amplificados fueron digeridos de acuerdo con las condiciones de reacción establecidas. Para asegurar la calidad de la validación se realizó un tamizaje al azar en el 10% de las muestras y se tamizaron aquellas con patrón homocigoto para alelos mutantes6.
Definición de eventos cardiovasculares adversos mayoresLa definición de isquemia recurrente, reinfarto, choque cardiogénico y defunción cardiovascular ha sido previamente publicada3. En resumen, para isquemia, nuevo dolor torácico con perfil isquémico y cambios dinámicos del ST (elevación o desnivel) en 2 derivaciones contiguas; para reinfarto, necrosis por electrocardiograma y biomarcadores; y para oclusión de la arteria relacionada, demostración angiográfica. Para choque cardiogénico se utilizaron variables clínicas, uso de vasopresores y datos hemodinámicos. Se consideró defunción cardiovascular toda mortalidad atribuida al evento índice de inestabilidad coronaria3.
SeguimientoLlamadas telefónicas y/o visita de consultorio a los 3, 6, 9 y 12 meses.
Análisis estadísticoSe evaluó la diferencia entre medias de grupos y variables continuas mediante t de Student de 2 colas; los resultados se verificaron con la prueba no paramétrica de la suma del rango de Wilcoxon. Se recogieron las variables discretas mediante chi-cuadrado con corrección de Yates o prueba exacta de Fisher. Se usaron razón de momios (RM) y riesgo relativo con intervalos de confianza (IC) del 95% (IC 95%) para determinar probabilidad, riesgo de enfermedad y ECAM. Se utilizaron modelos univariados de regresión logística y multivariada para identificar asociaciones entre variables; y Kaplan-Meier para ECAM (mortalidad, isquemia recurrente, reinfarto, choque cardiogénico y mortalidad) y supervivencia. Significación estadística: p<0,05. Los datos se miden en porcentajes, medias, desviaciones estándar, IC y RM. Se utilizó el paquete estadístico GBSTAT versión 10.0 of Dynamic Microsystems, Inc., Copyright 2004.
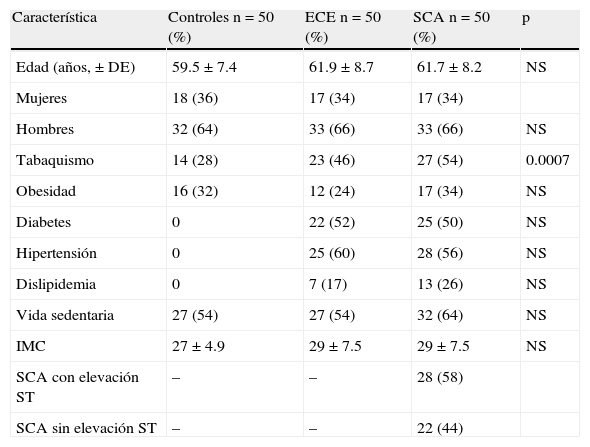
ResultadosDe septiembre del 2003 a agosto del 2005, 150 sujetos ingresaron al estudio. Las características demográficas de los 3 grupos se observan en la tabla 2. El promedio de edad fue alrededor de los 60 años con predominio de hombres. El tabaquismo fue una variable dominante en los pacientes con ECE y SCA en relación con los controles. Aunque estos se encontraban libres de enfermedad cardiovascular se observó tabaquismo, obesidad y sedentarismo en proporción similar a los otros grupos. La presencia de SCA con y sin elevación del ST fue similar en EC inestable. Angiográficamente en todos se demostró aterotrombosis. No se tuvo acceso a ultrasonido intracoronario.
Características demográficas
| Característica | Controles n=50 (%) | ECE n=50 (%) | SCA n=50 (%) | p |
| Edad (años,±DE) | 59.5±7.4 | 61.9±8.7 | 61.7±8.2 | NS |
| Mujeres | 18 (36) | 17 (34) | 17 (34) | |
| Hombres | 32 (64) | 33 (66) | 33 (66) | NS |
| Tabaquismo | 14 (28) | 23 (46) | 27 (54) | 0.0007 |
| Obesidad | 16 (32) | 12 (24) | 17 (34) | NS |
| Diabetes | 0 | 22 (52) | 25 (50) | NS |
| Hipertensión | 0 | 25 (60) | 28 (56) | NS |
| Dislipidemia | 0 | 7 (17) | 13 (26) | NS |
| Vida sedentaria | 27 (54) | 27 (54) | 32 (64) | NS |
| IMC | 27±4.9 | 29±7.5 | 29±7.5 | NS |
| SCA con elevación ST | – | – | 28 (58) | |
| SCA sin elevación ST | – | – | 22 (44) |
ECE: enfermedad coronaria estable; IMC: índice de masa corporal; NS: no significativo; SCA: síndromes coronarios agudos.
Se analizaron 14 polimorfismos en 9 genes. Fibrinógeno: 5 polimorfismos, 4 en el gen de la cadena β (−455 G/A, −148C/T, +1689 T/Gy Bcl-1) y uno en la cadena α (Taq I)⋅ Factor v: G1691A. Factor vii: R353Q. Factor ii: G20210A. Factor xiii: V34L. Inhibidor del activador del plasminógeno- 1 (IAP-1): 4G/5G. Para el gen del activador tisular del plasminógeno (ATP): AluI/D. PLA1/PLA2: glucoproteína IIIa, PLA1/PLA2. Proteína C reactiva: G1059C y el −717A/G.
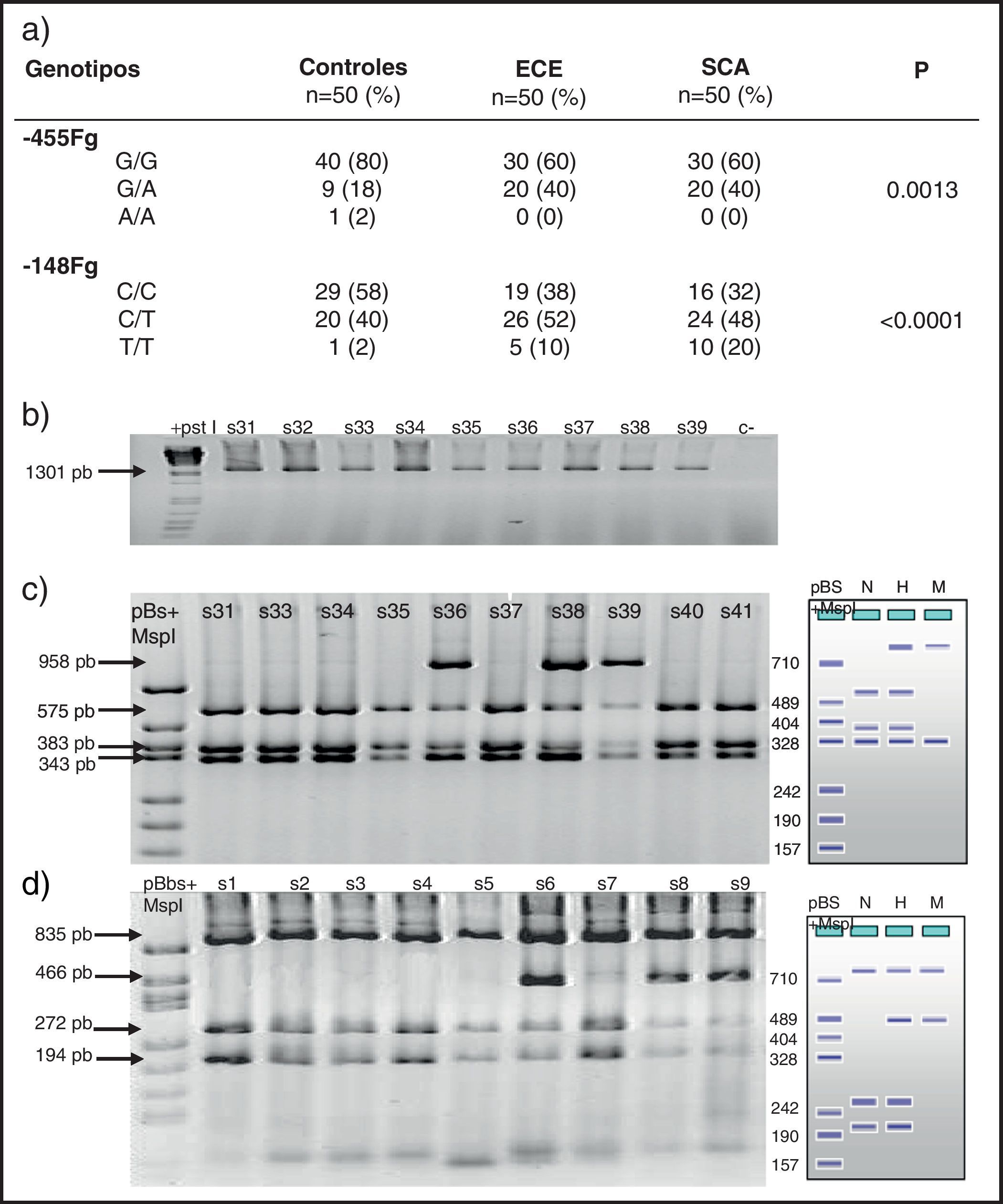
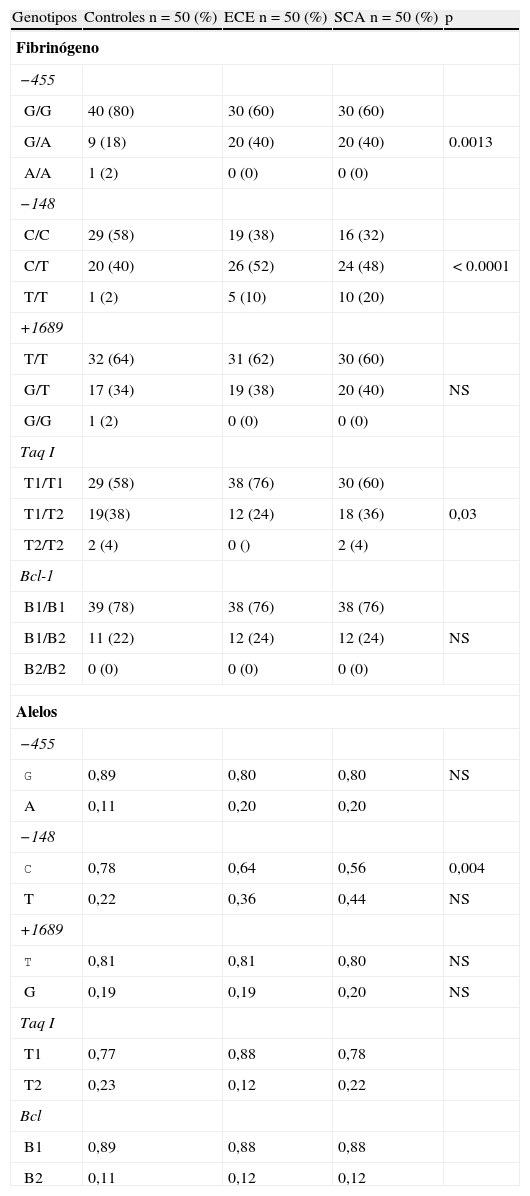
Frecuencias de polimorfismos en las 3 cohortesUn análisis preliminar de distribución de genotipos para cada polimorfismo no demostró desviaciones en la ley de equilibrio de Hardy-Weinberg (p>0,05). De los 4 estudiados, solo los localizados en el promotor del gen del fibrinógeno (−455 G/A y −148 C/T) y Taq I (gen fibrinógeno α) tuvieron diferencia estadísticamente significativa al comparar SCA, ECE y controles (tabla 3). La frecuencia de los genotipos −455 G/A y −48 C/T fue más significativa en los grupos con ECE o inestable (p=0,001 y p<0.0001). Sin embargo, al analizar los alelos solo la variante −148 T tuvo significación estadística (p=0,004). El resto no mostraron valores significativos para una condición clínica particular.
Genotipos del fibrinógeno en los grupos de estudio
| Genotipos | Controles n=50 (%) | ECE n=50 (%) | SCA n=50 (%) | p |
| Fibrinógeno | ||||
| −455 | ||||
| G/G | 40 (80) | 30 (60) | 30 (60) | |
| G/A | 9 (18) | 20 (40) | 20 (40) | 0.0013 |
| A/A | 1 (2) | 0 (0) | 0 (0) | |
| −148 | ||||
| C/C | 29 (58) | 19 (38) | 16 (32) | |
| C/T | 20 (40) | 26 (52) | 24 (48) | <0.0001 |
| T/T | 1 (2) | 5 (10) | 10 (20) | |
| +1689 | ||||
| T/T | 32 (64) | 31 (62) | 30 (60) | |
| G/T | 17 (34) | 19 (38) | 20 (40) | NS |
| G/G | 1 (2) | 0 (0) | 0 (0) | |
| Taq I | ||||
| T1/T1 | 29 (58) | 38 (76) | 30 (60) | |
| T1/T2 | 19(38) | 12 (24) | 18 (36) | 0,03 |
| T2/T2 | 2 (4) | 0 () | 2 (4) | |
| Bcl-1 | ||||
| B1/B1 | 39 (78) | 38 (76) | 38 (76) | |
| B1/B2 | 11 (22) | 12 (24) | 12 (24) | NS |
| B2/B2 | 0 (0) | 0 (0) | 0 (0) | |
| Alelos | ||||
| −455 | ||||
| G | 0,89 | 0,80 | 0,80 | NS |
| A | 0,11 | 0,20 | 0,20 | |
| −148 | ||||
| C | 0,78 | 0,64 | 0,56 | 0,004 |
| T | 0,22 | 0,36 | 0,44 | NS |
| +1689 | ||||
| T | 0,81 | 0,81 | 0,80 | NS |
| G | 0,19 | 0,19 | 0,20 | NS |
| Taq I | ||||
| T1 | 0,77 | 0,88 | 0,78 | |
| T2 | 0,23 | 0,12 | 0,22 | |
| Bcl | ||||
| B1 | 0,89 | 0,88 | 0,88 | |
| B2 | 0,11 | 0,12 | 0,12 | |
ECE: enfermedad coronaria estable; NS: no significativo; SCA: síndromes coronarios agudos.
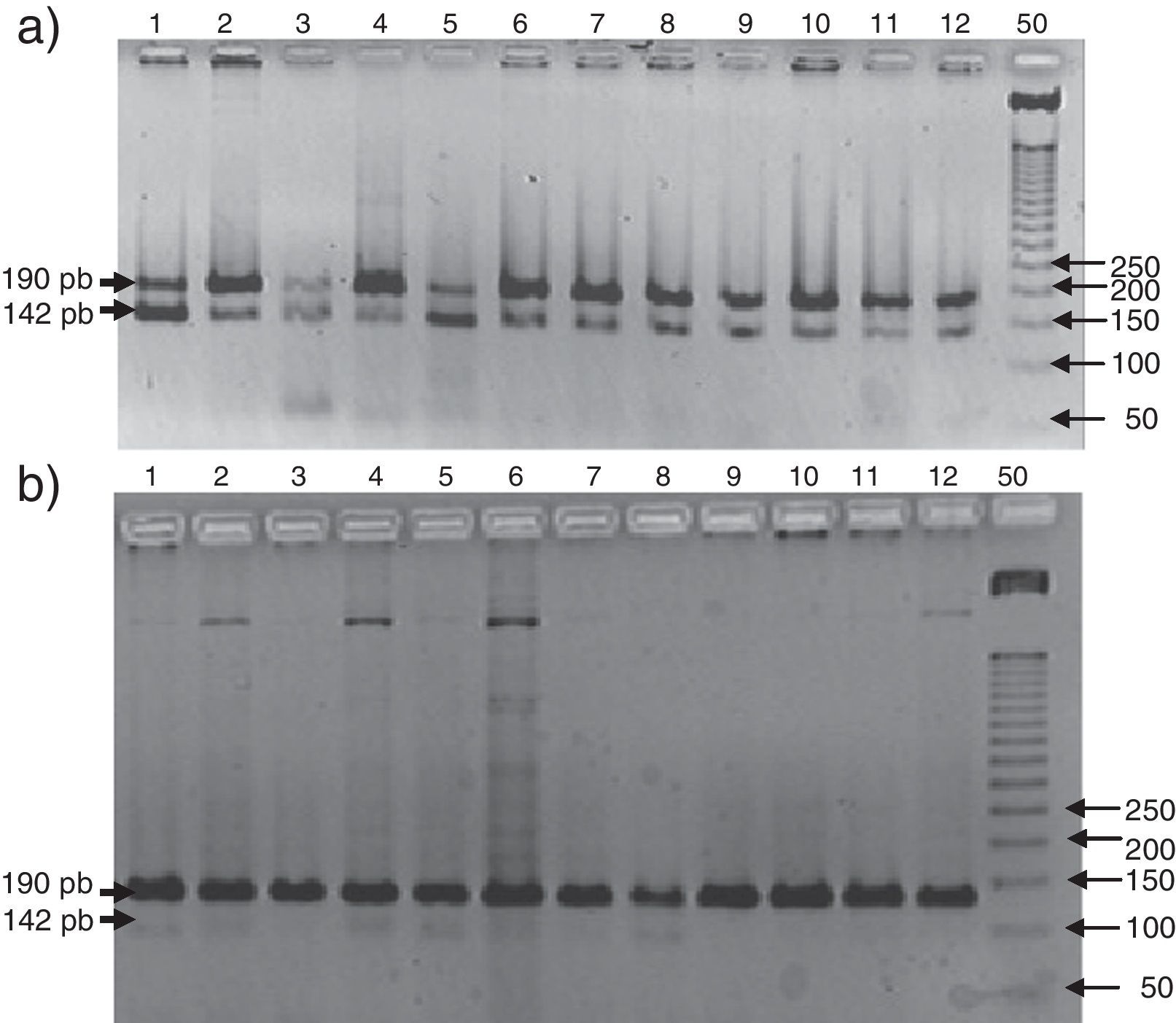
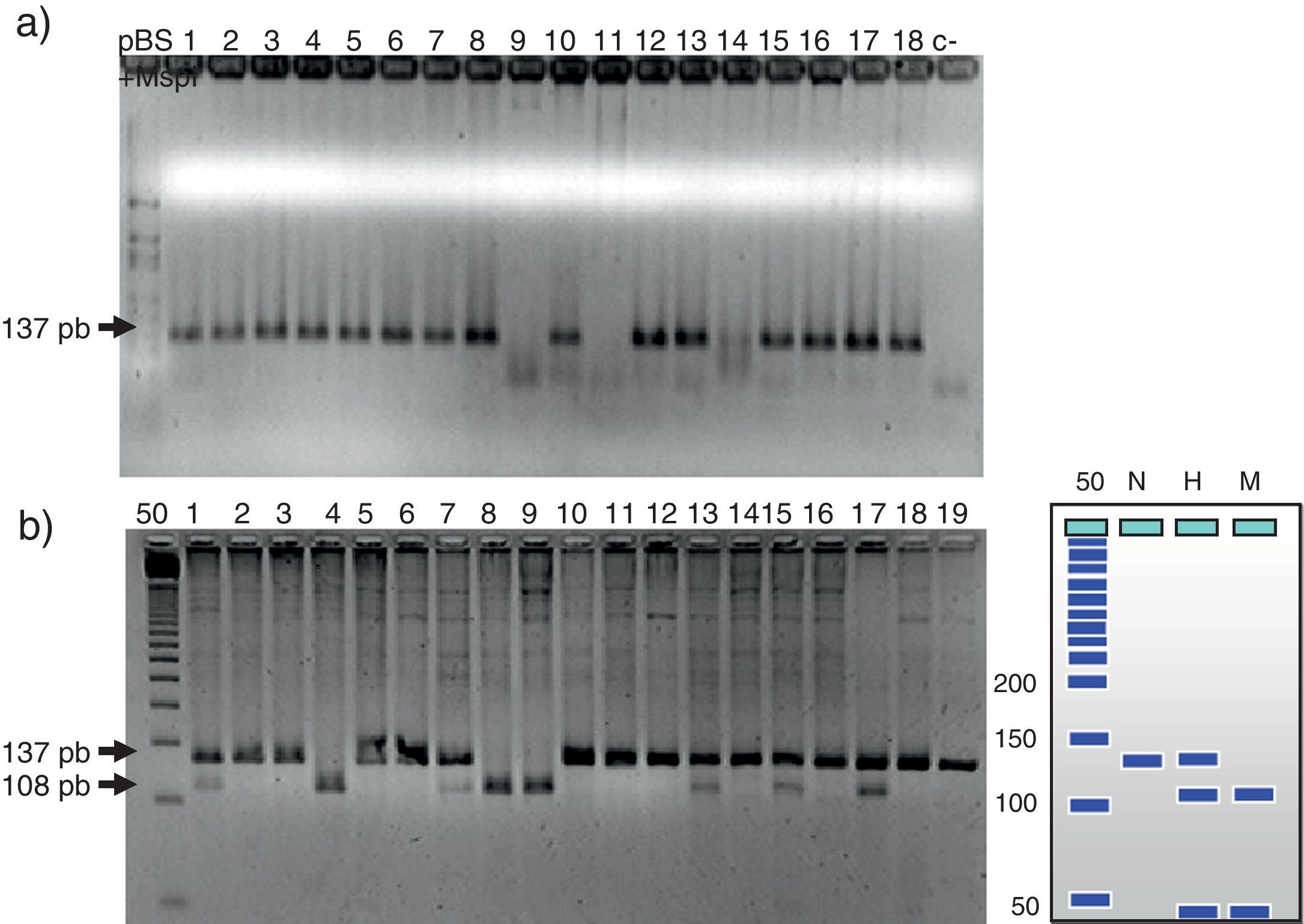
Al analizar la diferencia de frecuencias entre grupos solo los polimorfismos R353Q y V34L demostraron significación (p=0,03 y p<0.0001). Esta misma comparación con alelos sostuvo al V34L con significación (p<0.0001). Al analizar la diferencia de frecuencia entre grupos, solo los polimorfismo Alu D/I y −717 A/G tuvieron significación estadística (p=0,008 y 0.0002) y al compararlos con los alelos solo el Alu D/I permaneció estadísticamente significativo.
La determinación de las cargas genéticas de un alelo en particular entre los genotipos estudiados demostró incremento para EC. También se observó cómo la frecuencia de la carga del alelo −455A tuvo significado (RM 2.66, IC 95% 1.1-6.3, p=0,03) para las 2 condiciones patológicas. La carga del alelo −148T fue significativo para el grupo con ECE (RM 2.25, IC 95% 1.0-4.9, p=0,04) y para SCA (RM 2.93, IC 95% 1.3-6.4, p=0,01). El alelo −717 G solo tuvo significación en sanos y SCA (RM 9.33, p=0,03).
Variables bioquímicasLas variables bioquímicas con mayor significado estadístico entre sanos y ECE fueron: fibrinógeno (371.2+97.4 vs. 414.6+94.3mg/dL, p0,03), leucocitos (6.212+1.543 vs. 7.181+2.113 cél/mm3, p 0,01), neutrófilos (3.863+1.344 vs. 4.635+1.623 cél/mm3, p 0,01), creatinina (0,87+0,2 vs. 1.0+0,34mg/dL, p0,02).
Actividad de factores: factor ii (103.1+26.4 vs. 80.9+21.5%, p<0,001), factor v (97.2+33.1 vs. 72+26.7%, p0,001) y factor vii (122.9+32.0 vs. 93.8+33.4%, p<0,001).
Al comparar controles con SCA sostuvieron su significado estadístico: fibrinógeno (371.2+97.4 vs. 442.5+153.2.3mg/dL, p 0,007); leucocitos (6.212+1.543 vs. 8.982+3.244 cél/mm3, p<0,001); neutrófilos (3.863+1.344 vs. 6.727+2.932 cél/mm3, p<0.0001); creatinina (0,87+0,2 vs. 1.1+0,63mg/dL, p 0,02) y la actividad del factor ii (103.1+26.4 vs. 83.8+24.8%, p 0.0003).
Al concertar ECE con SCA mantuvieron significación: leucocitos (71.781+2.113 vs. 8.982+3.244 cél/mm3, p0,001) y neutrófilos (4.635+1.623 vs. 6.727+2.932 cél/mm3, p<0.0001) y los factores v (72+26.7 vs. 84.7+29.9%) y vii (93.8+33.4 vs 140.1+97.5%). Aunque el fibrinógeno no demostró ninguna diferencia (414.6+94.3 vs. 442.4+153.2mg/dL, p0,30), las cifras observadas (>350mg/dL) han demostrado ser una variable independiente de riesgo cardiovascular4.
Relación entre polimorfismos y niveles plasmáticos del fibrinógenoEl polimorfismo Bcl-1 (B1B2) se observó en el grupo con niveles entre 250 y 350mg/dL en el 14%; con un nivel entre 350 y 450mg/dL en el 24% y >450mg/dL en el 34% (p0,004). El polimorfismo −455Fg (G/A) se identificó en el 14, 36 y 50% respectivamente (p<0,001). El −148Fg (C/T) en el 46, 49 y 43% (0,001). El Tap1Fg (T1/T2) en el 33, 33 y 34% (0,06). El polimorfismo+1689Fg en un 26, 38 y 52% (0.0015). Los modelos de regresión múltiple que incluyeron variables históricas para ECAM (tabaquismo, edad, sexo, diabetes, dislipidemia, etcétera) tuvieron la mayor correlación cuando se asociaron los polimorfismos del fibrinógeno −455 y −148 (r=0,3, p=0,008).
En la figura 1 se observa en las 3 cohortes la distribución de los 2 polimorfismos del fibrinógeno con mayor impacto estadístico. El polimorfismo del factor viii se puede analizar en los 3 grupos en la figura 2. La figura 3 muestra la distribución genotípica del polimorfismo de la PCR (−717 A/G) en las 3 cohortes de pacientes.

En la parte izquierda se encuentra el esquema del gel de agarosa del polimorfismo −455 G/A con el marcador PM y en el carril N se encuentra representado el homocigoto G/G, en el carril H el heterocigoto G/A y en el M el homocigoto AA. A la derecha un gel de agarosa del polimorfismo −148C/T y como las muestras s31, s33, s34, s35, s37, s40 y s41 son homocigotas G/G y las muestras s36, s38 y s39 son heterocigotas G/A para el polimorfismo, −455.

 en las 3 cohortes de pacientes.")
La mayor incidencia de isquemia recurrente, reinfarto, choque y mortalidad se observó en la fase aguda. Isquemia recurrente: hospitalaria 10%, 3 meses 8%, 6 meses 5%, 9 meses 6% y 12 meses 5%. Reinfarto: hospitalario 2%, 3 meses 3% y 9 meses 1%. Choque: hospitalario 3%. Mortalidad: hospitalaria 3%, 3 meses de seguimiento 1%.
Correlación entre variables bioquímicas, hematológicas y polimorfismosSeguimiento a un año. En el grupo con SCA los niveles plasmáticos de fibrinógeno >450mg/dL (p=0,03) y leucocitos >8.500 cél/mm3(p=0,05), fueron marcadores de mal pronóstico. Se identificó la carga genética −148 CT/TT del fibrinógeno (RM 2.0, p0,04) y el −717 AG/GG de la PCR (RM 2.0, p0,04) como marcadores de isquemia recurrente. Para reinfarto se asoció el polimorfismo 1691GA+AA (RM 2.4, p0,01). Los neutrófilos fueron una variable independiente para choque cardiogénico (RM 2.0, p0,04) y la disfunción del ventrículo derecho para mortalidad (RM 1.9, p 0,05).
El modelo de regresión multivariado para isquemia recurrente (p=0,002) fue dominantemente genético e incluyó angiografía coronaria (RM 2.12, p 0,03), −148 CT+TT (RM 2.03, p0,04), PLA1/PLA2+PLA2/PLA2 (RM 2.28, p0,02). A un año de seguimiento, el modelo de regresión multivariado con mayor poder estadístico para mortalidad incluyó variables históricas, marcadores indirectos de disfunción endotelial por inflamación y el genotipo Alu I/I (p0,001). Las variables con significación estadística fueron fracción de expulsión (RM 1.96, p0,04) y el Alu I/I (RM 2.16, p0,03). El modelo de regresión multivariado para ECAM a un año de seguimiento (p=0,04) incluyó variables clínicas como hipertensión, angiografía coronaria, lesiones críticas y 2 cargas genotípicas en donde el polimorfismo −148 CT/TT del fibrinógeno fue el más significativo (RM 1.91, p0,05).
SeguimientoSe realizó seguimiento hospitalario a los 3, 6, 9 y 12 meses a través de visita médica y/o contacto telefónico. Esto se logró en el 97% de los casos. En los primeros 9 meses se perdió el contacto con 2 pacientes y a los 12 meses con otros 2.
DiscusiónEste estudio tiene 3 importantes hallazgos. Primero, la distribución genotípica de los genes sugiere una relación entre polimorfismos del fibrinógeno y de la hemostasia como marcadores de ECAM y de EC. Segundo, propone que la hiperfibrinogenemia que demostramos en diferentes modelos2,3,7 parece tener una predisposición genética2,3 más que ambiental4. Tercero, en nuestro medio y en diferentes estadios de EC4, se confirma el valor predictivo de leucocitos y fibrinógeno2,3,7,8.
En México la necesidad de estudios genéticos surge por las crecientes tasas de morbimortalidad e incapacidad por enfermedad vascular9–12, la variabilidad genética demostrada en diferentes grupos étnicos, la necesidad de identificar un perfil de susceptibilidad génica y porque en el futuro este puede ser el camino para identificar riesgo vascular, respuesta o nuevos objetivos terapéuticos y establecer efectivas estrategias de prevención primaria.
Enfermedad coronaria y genéticaEn la década pasada la aterosclerosis dejó de ser un padecimiento por depósito de colesterol y hoy se acepta como una enfermedad inflamatoria con una compleja interacción entre medio ambiente, disfunción endotelial y mensajes moleculares1,2. Además, las evidencias que demuestran que el endotelio va más allá de los vasa vasorum13, el concepto de remodelación vascular como expresión biológica de placas no estenóticas1,2, ruptura múltiple14,15 y la participación de factores de riesgo aterotrombóticos en la enfermedad tromboembólica venosa pulmonar16, le confieren una expresión de enfermedad sistémica, por lo que el concepto de «paciente vulnerable» debe extenderse al de «endotelio y sistema vascular vulnerable»16.
A través de avances en biología molecular se han detectado numerosos polimorfismos y mutaciones que al establecer un sinergismo con factores ambientales podrían estar implicados no solo en la génesis de la aterosclerosis (inflamación)17 sino también en algunos mecanismos íntimamente relacionados (metabolismo de los lípidos, resistencia a insulina, etcétera) con inestabilidad de la placa (actividad y reclutamiento de células inflamatorias y trombosis)17. Todo esto confiere a la EC un carácter poligénico y multifactorial, y establece la susceptibilidad genética como un factor de riesgo tan importante como los factores tradicionales.
Distribuciones genotípicas y polimorfismosLa distribución de los genes, además de establecer un vínculo entre el conocimiento biomolecular y los SCA12, podría ser –hasta nuestro conocimiento– la primera evidencia reportada en el país. Una aportación importante fue demostrar la ausencia de polimorfismos de riesgo cardiovascular observados en poblaciones caucásicas (G20210A del factor ii y G1691A del factor v)18–22.
Otro resultado importante fue identificar una relación entre polimorfismos de inflamación (fibrinógeno −148T) y coagulación (factores xiii V34L, ATP Alu I/D y PLA2 del GIIIa) con ECAM como expresión de un posible mecanismo multifactorial similar al observado con marcadores bioquímicos3. La misma expresión genética e impacto clínico se extendió en el seguimiento en donde polimorfismos de la misma clase (fibrinógeno −148Fg, −455 G/A y factor xiii V34L) también se relacionaron con EC.
Los polimorfismos en el promotor del fibrinógeno se vincularon con el fenotipo por una asociación independiente con los factores de riesgo históricos. Estos niveles de la proteína fueron superiores a otras evidencias23,24. En todo caso, las diferencias estadísticas observadas entre los grupos sugieren estratificaciones fenotípicas que pudieran relacionarse con un mayor riesgo cardiovascular.
Alelos −455 A y −148 TAl comparar sus cargas genéticas se observó relación con EC, lo que sugiere que los polimorfismos localizados en el promotor de la cadena β son responsables de regular los niveles de fibrinógeno asociados con enfermedad. El alelo −455 A en diferentes grupos étnicos25–28 ha demostrado una relación inconsistente con cardiopatía isquémica29,30 y el −148 T tiene una evidencia limitada como riesgo cardiovascular en poblaciones asiáticas y anglosajonas con infarto31 o enfermedad arterial periférica32. Por ello, esta evidencia podría ser la primera en establecer una relación entre estos alelos y EC en mexicanos.
En la fase aguda (alelo −455 A) y en el seguimiento (alelo −148 T) se observó una relación estrecha con valores anormales de fibrinógeno y ECAM, por lo que el alelo −148 T podría ser un marcador de EC. La asociación de ambos sugiere la posibilidad de un desequilibrio de ligamiento entre estos polimorfismos, sin que esto pueda establecerse por completo33,34. Su localización estratégica en la región promotora del gen del fibrinógeno β cerca del elemento de respuesta de la interleucina 6 y C/EBP, y de los sitios HNF1 y HNF327,35–39, es esencial para una respuesta completa del promotor a esta interleucina responsable de la respuesta inflamatoria y de niveles elevados de reactantes de fase aguda (fibrinógeno y proteína C). Su relación con ECAM podría atribuirse a una mayor respuesta del promotor del fibrinógeno a diferentes estímulos que generan un estado de disfunción endotelial agudo.
Alelo V34LEn este polimorfismo del factor xiii el cambio de aminoácido se encuentra muy cerca del anclaje de la trombina lo que podría ser muy importante en la activación de este factor. In vitro la 34 leucina parece formar trombos con mayor estabilidad a la fibrinólisis endógena40 sin embargo, otras evidencias sugieren que se asocia a trombo-resistencia e infarto41,42. Esto podría atribuirse a la interacción entre el factor xiii y la estructura de la fibrina con la actividad del fibrinógeno. Su alta concentración en muestras homocitogos para Leu se relaciona con trombos con mayor permeabilidad y una estructura más laxa con fibras densas que en aquellos formados por las muestras homocigotos Val. Por lo tanto el factor xiii en presencia de niveles de fibrinógeno relacionados con riesgo cardiovascular4 podría generar un mecanismo de trombo-resistencia a la fibrinólisis endógena y exógena con todas sus implicaciones clínicas.
Alelo PLA2 del GPIIIaSu importancia como marcador de isquemia recurrente se establece por su localización en el sitio de unión del fibrinógeno y la relación que parece existir entre inflamación y mecanismos de agregación plaquetaria43; esto sugiere que este alelo incrementa la actividad plaquetaria por una interacción con los niveles del fibrinógeno y mayor trombogénesis, y es una evidencia más de la interacción biológica entre plaquetas, leucocitos, trombosis e inflamación43.
Alelo Alu I/IAunque evidencias previas no lo han correlacionado con el ATP44–46, este alelo fue un marcador de mortalidad lo que sugiere que una hipoactividad de la fibrinólisis asociada a una reducción de sus niveles plasmáticos puede ser un factor de riesgo para trombosis. La naturaleza de inserción de una repetición Alu en un intrón hace difícil que esta mutación afecte los niveles de esta proteína; sin embargo, es posible alterar la estabilidad del mARN47 considerando que las concentraciones del ATP dependen de su secreción y separación, así como del complejo que forma con el IAP-148.
Por otra parte, la determinación del ATP secretado por células endoteliales supera a la determinación de niveles circulantes49. Posterior a su expresión endotelial pierde rápidamente actividad por la separación mediada por receptores del hígado y la interacción del IAP-1. Su mayor actividad lítica sobre la fibrina se observa en el trombo formado50,51 y cuando su secreción en las células endoteliales es mayor que los niveles circulantes. Datos recientes apoyan la relación entre el polimorfismo Alu I/D y la secreción del ATP52,53 lo que sugiere que este alelo se asocia con un sistema fibrinolítico anormal y trombogénesis.
Leucocitos y fibrinógenoComo hemos demostrado previamente3,7, ambos marcadores indirectos de disfunción endotelial e inflamación fueron marcadores de mal pronóstico. El mecanismo parece ser multifactorial. Los leucocitos se asocian a hipercoagulabilidad, fenómeno de no-reflujo, interleucinas y moléculas de adhesión celular-1. El fibrinógeno participa en la formación de trombina, agregación plaquetaria, modula la disfunción endotelial, promueve proliferación y migración de las células del músculo liso, interactúa con las uniones de plasmina y es una proteína mayor de fase aguda4. Los alelos del fibrinógeno demostrados sugieren una posible participación genética más que ambiental4. Si ambos se encuentran involucrados o no en la causalidad de la aterotrombosis y en la génesis de ECAM, está aún por determinarse. Sin embargo, mientras esta interrogante y otras esperan una respuesta, estos indicadores de inflamación emergen como promisorios marcadores adicionales de riesgo cardiovascular que podrían estratificar el riesgo en diferentes estadios de la EC.
Implicaciones clínicasLos marcadores genéticos y bioquímicos demostrados son una evidencia más de la compleja interacción entre medio ambiente, inflamación, disfunción endotelial y mensajes moleculares. La evidencia actual establece al fibrinógeno4 como un factor fuerte, consistente e independiente o un indicador de riesgo, y podría ser el vínculo perdido entre la enfermedad cardiovascular y los factores clásicos de riesgo. Además, por su participación activa en mecanismos de resistencia o respuesta subóptima a heparina4, terapia fibrinolítica4 e inhibidores de los receptores de superficie plaquetaria IIb/IIIa54, podría ser un importante predictor de respuesta terapéutica. Estos resultados podrían considerarse como un estudio piloto para generar un estudio representativo en población mexicana y mejorar su validez interna y externa.
LimitacionesMuestra y población limitada, por el costo de los recursos para este tipo de estudios, lo que reduce la validez interna y externa de los resultados; se incluyeron pacientes con SCA con y sin elevación del ST en una pequeña población del noreste de México.
ConclusiónEn pacientes con un SCA se demostró una relación entre polimorfismos relacionados con hemostasia e inflamación con ECMA, por lo que podrían considerarse marcadores de enfermedad coronaria. Se requiere una muestra mayor para confirmar estos resultados.
FinanciaciónEl estudio fue aprobado y recibió financiamiento por el Fondo Sectorial de Investigación en Salud y Seguridad Social en la convocatoria SSA/IMSS/ISSTE-CONACYT 2002 (SALUD-2002-CO1-7162).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.