




The American Thoracic Society and the Infectious Diseases Society of America recommend that clinically significant non-tuberculous mycobacteria (NTM) should be identified to the species level in order to determine their clinical significance. The aim of this study was to evaluate identification of rapidly growing NTM (RGM) isolated from clinical samples by using MALDI-TOF MS and a commercial molecular system. The results were compared with identification using a reference method.
MethodsWe included 46 clinical isolates of RGM and identified them using the commercial molecular system GenoType® CM/AS (Hain, Lifescience, Germany), MALDI-TOF MS (Bruker) and, as reference method, partial rpoβ gene sequencing followed by BLAST and phylogenetic analysis with the 1093 sequences available in the GeneBank.
ResultsThe degree of agreement between GenoType® and MALDI-TOF MS and the reference method, partial rpoβ sequencing, was 27/43 (62.8%) and 38/43 cases (88.3%) respectively. For all the samples correctly classified by GenoType®, we obtained the same result with MALDI-TOF MS (27/27). However, MALDI-TOF MS also correctly identified 68.75% (11/16) of the samples that GenoType® had misclassified (p=0.005).
ConclusionsMALDI-TOF MS classified significantly better than GenoType®. When a MALDI-TOF MS score >1.85 was achieved, MALDI-TOF MS and partial rpoβ gene sequencing were equivalent. GenoType® was not able to distinguish between species belonging to the M. fortuitum complex. MALDI-TOF MS methodology is simple, rapid and associated with lower consumable costs than GenoType®. The partial rpoβ sequencing methods with BLAST and phylogenetic analysis were not able to identify some RGM unequivocally. Therefore, sequencing of additional regions would be indicated in these cases.
La American Thoracic Society y la Infectious Diseases Society of America recomiendan que las micobacterias no tuberculosas (MNT) clínicamente relevantes sean identificadas a nivel de especie para determinar su significado clínico. El propósito de este estudio fue a partir de MNT de crecimiento rápido (MCR) aisladas en muestras clínicas, evaluar su identificación mediante MALDI-TOF MS y un método molecular comercial, comparando estos resultados con la identificación obtenida usando un método de referencia.
MétodosSe incluyeron 46 aislados clínicos de MCR. Estos aislados se identificaron mediante el método molecular comercial GenoType® Mycobacterium CM/AS (Hain, Lifescience, Alemania), MALDI-TOF MS (Bruker) y, como método de referencia, la secuenciación parcial del gen rpoβ seguido de BLAST y análisis filogenético. Para el análisis filogenético se utilizaron 1.093 secuencias disponibles en el GeneBank.
ResultadosEntre GenoType® o MALDI-TOF MS, la concordancia respecto al método de referencia, secuenciación parcial de rpoB, fue 27/43 (62,8%) y 38/43 casos (88,3%), respectivamente. En todas las muestras que GenoType® clasificó correctamente con MALDI-TOF MS se obtuvo el mismo resultado (27/27). Pero además MALDI-TOF MS identificó bien 68,75% (11/16) de las muestras que GenoType® no clasificó correctamente (p=0,005).
ConclusionesMALDI-TOF MS clasificó significativamente mejor que GenoType®. Cuando MALDI-TOF MS alcanzó una puntuación >1,85, MALDI-TOF y la secuenciación parcial del gen rpoβ fueron equivalentes. GenoType® no distinguió dentro del M. fortuitum complex. La metodología MALDI-TOF MS es simple, rápida y se asocia a un menor coste de consumibles que GenoType®. La secuenciación parcial del gen rpoβ con BLAST y análisis filogenético no lograron identificar de manera inequívoca algunas MCR. Para estas MCR estaría indicado la secuenciación de regiones adicionales.
The last twenty years have been characterized by an extraordinary increase of new Mycobacterium spp. Among over 150 officially recognized nontuberculous mycobacteria (NTM), only a few tens are familiar to clinicians.1 NTM are widely present in the environment and commonly inhabit soil and water. This group of microorganisms are considered to be opportunistic pathogens which can cause pulmonary, skin, soft tissue, lymphatic and disseminated infections, as well as nosocomial outbreaks related to inadequate disinfection/sterilization of medical equipment. The incidence of diseases caused by NTM is on the rise due to, among other reasons, an increase in immunocompromised patients.2 The American Thoracic Society and the Infectious Diseases Society of America recommend that clinically significant NTM should be identified to the species level in order to determine their clinical significance and select appropriate treatments.3
Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) allows the identification of organisms on the basis of unique spectral fingerprints produced by extracted proteins. In recent years, MALDI-TOF MS has irrupted as a rapid, powerful and relatively inexpensive tool for the identification of bacteria and yeast in the clinical laboratory setting.1,4
The molecular approach, using both in-house and commercial systems, is the most frequently used method worldwide for the identification of mycobacterial species. In this work, two molecular approaches for mycobacterial identification are used: a molecular commercial system and partial RNA polymerase beta-subunit gene sequencing (rpoβ).5
Genetic sequencing of conserved genes is the referent method for the identification of mycobacteria.1 A number of targets useful for identification purposes have been detected within the genome of mycobacteria. Several studies have indicated that the hypervariable region of the rpoβ gene, located between 2300 and 3300bp, is required for suitable identification at species level and this region is recommended as a starting point for identification.6
The aim of this study was to evaluate rapidly growing mycobacteria (RGM) identification from clinical samples by using MALDI-TOF MS technology and a molecular commercial system, both methodologies are used in the routine practice of our laboratory. For this purpose, we compared the results obtained by using the routine methodology with the results obtained by using gold standard methodology (partial rpoβ gene sequencing phylogenetic analysis).
Material and methodsClinical isolatesFrom January 2007 to April 2015, a total of 46 clinical isolates of RGM were obtained from patients belonging to the health area of Santiago de Compostela (458.759 inhabitants), Galicia, Northwest of Spain. Only 15 of these patients presented diagnosis criteria according to the American Thoracic Society (ATS).3 These mycobacterial isolates were firstly identified by using the molecular commercial system GenoType® CM/AS, Hain Lifescience, Nehren, Germany. Clinical isolates were preserved at −20°C in skimmed milk until they were used. Thereafter, each strain was inoculated into agar blood medium (Columbia-blood agar base, Becton Dickinson, New Jersey, USA) and incubated at 37°C in a 10% CO2 incubator. After 5–7 days the growth achieved was enough to carry out the corresponding studies.
Clinical isolates were obtained as part of the routine activity of the Department of Microbiology and they were analyzed anonymously in a retrospective manner. Thus, ethical approval and informed consent were not required.
GenoType®Mycobacterium CM/AS (Hain, Lifescience, Germany) (GenoType®)The GenoType® includes a multiplex PCR followed by reverse hybridization and line probe technology. This assay targets the 23S rRNA gene and is available as two kits: CM, which identifies 22 of the most frequently isolated species, and AS, which identifies an additional 13 species. The GenoType® assay was performed according to the manufacturer's instructions. The final identification was obtained by comparison between the line probe patterns and the evaluation sheet provided by manufacturers.
MALDI-TOF MS protein extraction protocolIt was used the inactivated mycobacteria bead preparation method provided by Bruker Daltonics GmbH (Bremen, Germany). With a sterile 10μL loop, mycobacterial colonies growing on solid medium (agar blood) were transferred into a 1.5mL screw-cap microcentrifuge tub which contained 300μL of sterile water. Then, it was heated for 30min at 100°C in a thermoblock to inactivate the RGM. On average, colonies were 5–7 days old, depending on the growth rate of the mycobacteria. The tubes were allowed to cool at room temperature for 2min. Then, 900μL of 100% ethanol were added. After vortexing, the tubs were centrifuged at 13,000rpm for 2min and the supernatant was discarded. The tubes were centrifuged again at 13,000rpm for 2min and all residual liquid were completely removed. The pellet was allowed to dry at room temperature for 5min. The pellet was suspended in 25μL of acetonitrile, and approximately a volume of 50μL of 0.5mm diameter glass beds was added to the pellet. The tubes were vortexed for 1min, 25μL of 70% formic acid were added, and the samples were vortexed again for 1min. The tubes were centrifuged at 13,000rpm for 2min, and the supernatant was used for analysis with MALDI-TOF MS.
All steps requiring open manipulation of mycobacteria before heat inactivation were performed inside a biological safety cabinet in a biosafety level 2 setup.
MALDI-TOF MS analysisStrains were processed using a MALDI-TOF MS microflex LT (Brucker Daltonics GmbH, Bremen, Germany). Analyses were performed according to the manufacturer's instructions.
One microliter of supernatant was spotted in duplicate onto a steel target (MTP 384 polished steel target plate BC; Brucker Daltonics GmbH, Bremen, Germany) and air dried at room temperature. One microliter of matrix solution (saturate solution of alfa-cyano-4-hydroxycinnamic acid in 50% acetonitrile and 2.5% trifluoroacetic acid) was pipetted onto each of the spotted samples. After drying, the target was inserted into the MALDI-TOF MS. MALDI-TOF MS analysis was performed in automatic mode. Spectra were acquired at a laser frequency of 60Hz across a mass/charge ratio (m/z) of 2000–20,000Da.
The protein profile was obtained by the software FlexControl 3.4 (Brucker Daltonics GmbH, Bremen, Germany). Spectra were analyzed against those in the Brucker database Maldi Biotyper Mycobacteria Library 3.0 (853 references from 149 Mycobacterium species) using the Maldi Biotyper software (v 3.1; Bruker Daltonics). This assigned a logarithmic score ranging from 0 to 3 and determined the best match based on the m/z ratio and relative peak size of each ionized protein.
Partial rpoβ gene sequencingDNA extractionIt was performed using the DNA extraction method for solid culture included in the Anyplex™ MTB/NTM Real-time Detection V2.0 (Seegene, Seoul, Korea) according to the manufacturer's instructions.
rpoβ amplification5μL of extracted DNA were used for PCR in a 25μL final reaction volume by using the reagents provided by the GoTaq® PCR Core Systems I (Promega) kit: 5U/μL GoTaq® DNA Polymerase, 5X Colorless GoTaq® Flexi Buffer (Mg-Free), 25mM MgCl2 and 10mM PCR Nucleotide Mix. The direct and reverse primers were 10μM. The final concentrations were 0.025U/μL GoTaq® DNA Polymerase, 4mM MgCl2, 0.2mM dNTPs (each nucleotide), 0.2μM each primer. All the clinical strains were identified by partial rpoβ gene sequencing using a previously described method.5 Briefly, flanking the most variable rpoβ region, PCR primers MycoF (5′-GGCAAGGTCACCCCGAAGGG-3′; base positions 2573–2592, the nucleotides have been numbered according to the M. smegmatis ATCC 14468 rpoβ gene sequence) and MycoR (5′-AGCGGCTGCGGGTGATCATC-3′; base positions 3316–3337) amplified a 764-bp fragment from the rpoβ gene region in clinical isolates. Conditions for rpoβ gene amplification were pre-heated at 95°C for 10min, 35 cycles of 95°C for 30s, 55°C for 30s and 72°C for 90s; finally, an elongation step of 72°C for 7min. Five microliters of PCR product were analyzed by ultraviolet fluorescence after having been stained with ethidium bromide in a 1% agarose gel.
rpoβ partial sequencingPCR products were purified by using Qiagen columns (Qiagen, Germany). Purified amplicons were sequenced by using the GenomeLab™ Dye Terminator Cycle Sequencing with the Quick Start kit according to the manufacturer's instructions (Beckman Coulter®) with the following program: 30 cycles of denaturation at 96°C for 20s, primer annealing at 50°C for 20s and extension at 60°C for 4min. The sequencing primers were the same as in the amplification. Products of sequencing reactions were read in an automatic sequencer (CEQ™ 8000 Genetic Analysis System, Beckman Coulter®) by following the standard protocol of the supplier.
Those partial rpoβ gene sequences obtained in that way were compared to those available in the GeneBank using BLAST software (NCBI) (http://blast.ncbi.nim.nihgov/Blast.cgi-). After that, the most probable bacterial species were selected. Partial rpoβ gene sequences that displayed at least 98% sequence identity when compared to those in the GeneBank were considered as identified species.5
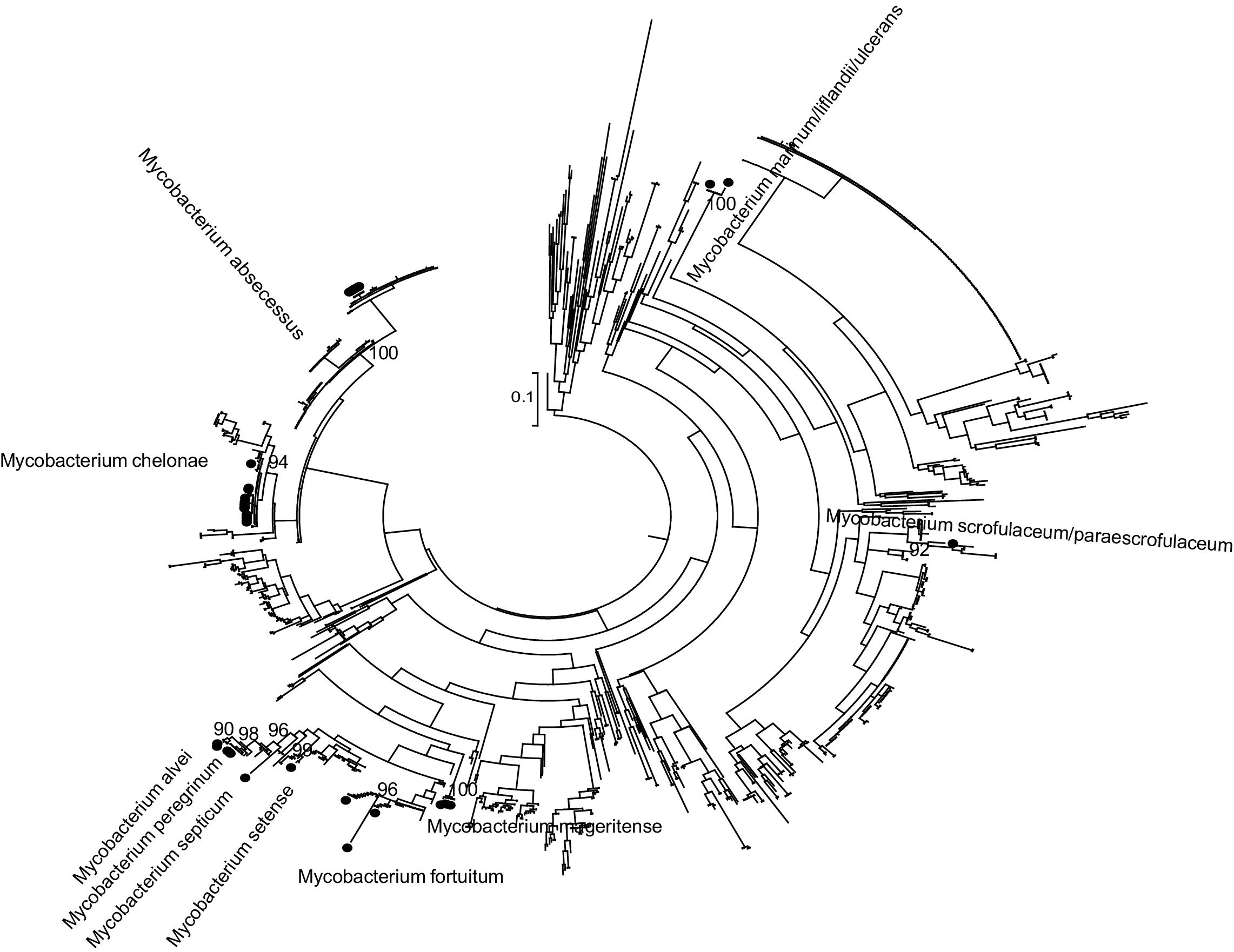
Phylogenetic analysisPhylogenetic analysis was carried out to improve the accuracy of partial rpoβ gene sequencing. The partial rpoβ gene sequence length ranged between 278 and 710 nucleotides. A set of reference sequences of Mycobacterium's rpoβ gene available in the GeneBank was obtained by using BLAST (Table S1 included as Supplementary material). The reference data set including 1093 sequences and nucleotide sequences were translated and aligned using the ClustalW algorithm implemented in MEGA. The sequences were divided according to the sequenced fragment for analysis. Phylogenetic trees were reconstructed by means of ML with PhyML7 3.0 by using the general time reversible plus proportion of invariable sites plus gamma distribution parameter (GTR) plus I plus G evolutionary model selected with jModeltest8 0.1 and a BIONJ starting tree. Heuristic tree searches under the ML optimality criterion were performed using the nearest neighbor interchange (NNI) branch-swapping algorithm. The approximate likelihood ratio test (aLRT) based on a Shimodaira–Hasegawa-like procedure was used as a statistical test to calculate branch support.9 Only aLRT support values of >90% were considered statistically significant and displayed at the tree nodes.
Statistical analysisThe Kappa coefficient was used to study the degree of concordance between methods. Fisher's exact test was used to evaluate the agreement between GenoType® and MALDI-TOF. A p value <0.05 was considered statistically significant. The analyses were done with STATA 13.1 (StataCorp, USA).
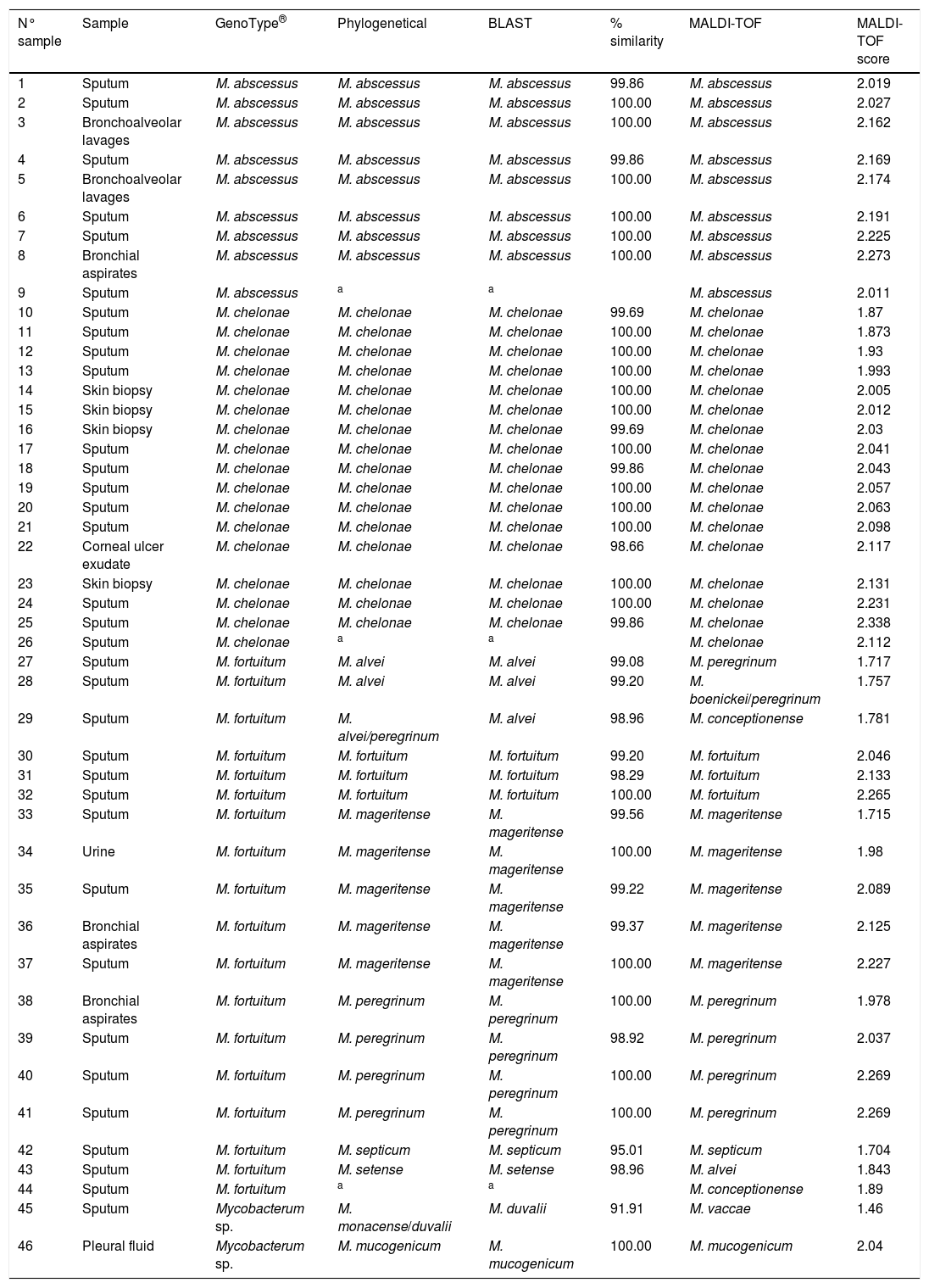
ResultsThe results obtained are shown in Table 1 and Fig. 1 (Figures 2–8 are included as Supplementary material).
Clininal samples, identification with GenoType®, phylogenetical analysis, BLAST, % similarity with BLAST, identification with MALDI-TOF and MALDI-TOF score.
| N° sample | Sample | GenoType® | Phylogenetical | BLAST | % similarity | MALDI-TOF | MALDI-TOF score |
|---|---|---|---|---|---|---|---|
| 1 | Sputum | M. abscessus | M. abscessus | M. abscessus | 99.86 | M. abscessus | 2.019 |
| 2 | Sputum | M. abscessus | M. abscessus | M. abscessus | 100.00 | M. abscessus | 2.027 |
| 3 | Bronchoalveolar lavages | M. abscessus | M. abscessus | M. abscessus | 100.00 | M. abscessus | 2.162 |
| 4 | Sputum | M. abscessus | M. abscessus | M. abscessus | 99.86 | M. abscessus | 2.169 |
| 5 | Bronchoalveolar lavages | M. abscessus | M. abscessus | M. abscessus | 100.00 | M. abscessus | 2.174 |
| 6 | Sputum | M. abscessus | M. abscessus | M. abscessus | 100.00 | M. abscessus | 2.191 |
| 7 | Sputum | M. abscessus | M. abscessus | M. abscessus | 100.00 | M. abscessus | 2.225 |
| 8 | Bronchial aspirates | M. abscessus | M. abscessus | M. abscessus | 100.00 | M. abscessus | 2.273 |
| 9 | Sputum | M. abscessus | a | a | M. abscessus | 2.011 | |
| 10 | Sputum | M. chelonae | M. chelonae | M. chelonae | 99.69 | M. chelonae | 1.87 |
| 11 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 1.873 |
| 12 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 1.93 |
| 13 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 1.993 |
| 14 | Skin biopsy | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.005 |
| 15 | Skin biopsy | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.012 |
| 16 | Skin biopsy | M. chelonae | M. chelonae | M. chelonae | 99.69 | M. chelonae | 2.03 |
| 17 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.041 |
| 18 | Sputum | M. chelonae | M. chelonae | M. chelonae | 99.86 | M. chelonae | 2.043 |
| 19 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.057 |
| 20 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.063 |
| 21 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.098 |
| 22 | Corneal ulcer exudate | M. chelonae | M. chelonae | M. chelonae | 98.66 | M. chelonae | 2.117 |
| 23 | Skin biopsy | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.131 |
| 24 | Sputum | M. chelonae | M. chelonae | M. chelonae | 100.00 | M. chelonae | 2.231 |
| 25 | Sputum | M. chelonae | M. chelonae | M. chelonae | 99.86 | M. chelonae | 2.338 |
| 26 | Sputum | M. chelonae | a | a | M. chelonae | 2.112 | |
| 27 | Sputum | M. fortuitum | M. alvei | M. alvei | 99.08 | M. peregrinum | 1.717 |
| 28 | Sputum | M. fortuitum | M. alvei | M. alvei | 99.20 | M. boenickei/peregrinum | 1.757 |
| 29 | Sputum | M. fortuitum | M. alvei/peregrinum | M. alvei | 98.96 | M. conceptionense | 1.781 |
| 30 | Sputum | M. fortuitum | M. fortuitum | M. fortuitum | 99.20 | M. fortuitum | 2.046 |
| 31 | Sputum | M. fortuitum | M. fortuitum | M. fortuitum | 98.29 | M. fortuitum | 2.133 |
| 32 | Sputum | M. fortuitum | M. fortuitum | M. fortuitum | 100.00 | M. fortuitum | 2.265 |
| 33 | Sputum | M. fortuitum | M. mageritense | M. mageritense | 99.56 | M. mageritense | 1.715 |
| 34 | Urine | M. fortuitum | M. mageritense | M. mageritense | 100.00 | M. mageritense | 1.98 |
| 35 | Sputum | M. fortuitum | M. mageritense | M. mageritense | 99.22 | M. mageritense | 2.089 |
| 36 | Bronchial aspirates | M. fortuitum | M. mageritense | M. mageritense | 99.37 | M. mageritense | 2.125 |
| 37 | Sputum | M. fortuitum | M. mageritense | M. mageritense | 100.00 | M. mageritense | 2.227 |
| 38 | Bronchial aspirates | M. fortuitum | M. peregrinum | M. peregrinum | 100.00 | M. peregrinum | 1.978 |
| 39 | Sputum | M. fortuitum | M. peregrinum | M. peregrinum | 98.92 | M. peregrinum | 2.037 |
| 40 | Sputum | M. fortuitum | M. peregrinum | M. peregrinum | 100.00 | M. peregrinum | 2.269 |
| 41 | Sputum | M. fortuitum | M. peregrinum | M. peregrinum | 100.00 | M. peregrinum | 2.269 |
| 42 | Sputum | M. fortuitum | M. septicum | M. septicum | 95.01 | M. septicum | 1.704 |
| 43 | Sputum | M. fortuitum | M. setense | M. setense | 98.96 | M. alvei | 1.843 |
| 44 | Sputum | M. fortuitum | a | a | M. conceptionense | 1.89 | |
| 45 | Sputum | Mycobacterum sp. | M. monacense/duvalii | M. duvalii | 91.91 | M. vaccae | 1.46 |
| 46 | Pleural fluid | Mycobacterum sp. | M. mucogenicum | M. mucogenicum | 100.00 | M. mucogenicum | 2.04 |
 Mycobacterium rpoβ partial gene 495 nucleotides.")
Phylogenetic and BLAST analyses were in agreement, but in 2 strains, phylogenetic analysis found no significant differences between at least two species of Mycobacterium spp. (Table 1: strains 29 and 45). Strains were considered correctly classified if any of these results were obtained.
By using BLAST, samples 42 and 45 displayed a sequence identity less than 98%, when they were compared to those available in the GeneBank. The identification was in accordance with phylogenetic analysis. For sample 45, phylogenetic analysis found no significant differences between M. duvalii and M. monacense.
Three strains (6.5%) were not identified using partial rpoβ gene sequencing (BLAST and phylogeny) because it was not possible to amplify them by PCR.
Eight strains were identified by partial rpoβ gene sequence as M. abscessus. It was not possible classified these M. abscessus at subspecies level.
GenoType® and partial rpoβ gene sequencingAgreement between GenoType® and partial rpoβ sequencing methods was observed in 27 of 43 cases (62.8%), Kappa value 0.534 (p<0.001).
MALDI-TOF MS and partial rpoβ gene sequencingAgreement between MALDI-TOF MS and partial rpoβ sequencing methods was observed in 38 of 43 cases (88.3%), Kappa value 0.854 (p<0.001).
When only MALDI-TOF scores >1.85 were considered, the agreement was 100% (36 of 36). For MALDI-TOF scores <1.85, only 2 of 7 (28.6%) results were in accordance (Table 1).
GenoType® and MALDI-TOF MSAll the strains correctly classified by GenoType® were also well identified when using MALDI-TOF MS (27/27). However, MALDI-TOF MS detected correctly a 68.75% (11/16) of the strains that GenoType® misclassified (p=0.005).
DiscussionIn this work, the identification of RGM using the routine methodology (GenoType® and MALDI-TOF MS) and a reference methodology (partial rpoβ gene sequencing and phylogenetic analysis) were compared.
Procedures for the identification of mycobacteria have greatly changed in the last years, and conventional biochemical and cultural methods are nowadays used only by a few laboratories worldwide. These methods have been abandoned because, in addition to relying on tests that are poorly reproducible and unbearably time consuming, they lack sufficient discriminative power (1). Even the HPLC methods based on analysis of cell wall lipids now seem to be in trouble because of the steady increment in the number of species within the genus Mycobacterium.10 In the last few years, the emergence of new species sharing common HPLC profiles has steadily diminished the discriminatory capacity of this method.1
There are some works which support MALDI-TOF MS technology to identify Mycobacterium spp.4,11 Clark et al. conclude that these studies demonstrate the feasibility and strong accuracy associated to MALDI-TOF MS for mycobacterial identification.4 Nevertheless, other authors believe that the accuracy of the identification achievable at present with genetic approaches is out of reach for MALDI-TOF MS technology.1 In this work, we have found that it is necessary to reach a specific MALDI-TOF MS score value (1.85) so that the identifications obtained with MALDI-TOF MS are correct with respect to the reference method12 which was used. However, it must be taken into account the limited number of isolates and varieties of RGM species which were used in our study.
From the point of view of cost-effectiveness, the results were obtained within 2h with MALDI-TOF MS while it took at least 4h with GenoType®. Moreover, MALDI-TOF MS methodology is simple, quick and associated with significantly lower consumable costs than GenoType®. The MALDI-TOF mass spectrometer and associated software are expensive initially but the continuing consumable costs are inexpensive (less than 1 $, 1.25 €, per isolate).11 The cost per identification with GenoType®GenoType in our laboratory goes up to 49 € each determination, which multiplies several times the cost for each determination obtained with MALDI-TOF MS.
M. abscessus complex comprises a group of multidrug-resistant RGM that has emerged as an important pathogen responsible for a wide spectrum of infections, following its recognition as a different entity from M. chelonae in 1992. In this work these three methods were able to differentiate these species with total accordance.
M. abscessus complex is differentiated into 3 subspecies.13 These subspecies have clinical relevance because they are related to the response to macrolides. M. abscessus subsp. bolletii infection had a higher response rate (approximately 90%) than patients with M. abscessus subsp. abscessus infection (approximately 25%).14 Most M. abscessus subsp. massiliense strains do not posses inducible macrolide resistance. In contrast with some authors15,16 and in agreement with others,17,18 we found that the partial rpoβ gene sequencing used in our study5 was unable to classify M. abscessus subspecies. It is widely accepted that several genes, analyzed in a particular way by researches, are necessary to identify the three subspecies: rpoβ, secA, sodA and hsp65 genes.17,18 The single gene-based approach is sensitive to the evolutionary history of that gene, but not necessarily of the species, and it may be limited by the lack of sufficient variation in a single gene sequence. Therefore, a multiple gene-based approach may lead to an accurate inference of phylogenetic tree for subspecies classification.19 Nevertheless some authors have described methods in which MALDI-TOF MS is used to differentiate M. abscessus complex at subsp. level.20 We consider that it would be of great interest to expand the study for the identification of this species of mycobacteria and its subspecies by means of MALDI-TOF MS due to its vital clinical importance.
M. fortuitum complex comprises the following species: M. fortuitum, M. peregrinum, M. senegalense, M. setense, M. mageritense, M. septicum, M. alvei, M. houstonense, M. boenickei, M. conceptionense, M. porcinum, M. neworleansense and M. brisbanense. In our study we found 18 M. fortuitum complex isolates but only six different species. According to the manufacturer's instructions, GenoType® differentiates M. fortuitum type 1, M. fortuitum type 2/M. mageritense and M. peregrinum/M. alvei/M. septicum but, in this study, all of them were identified as M. fortuitum by GenoType®. Despite the limitation implied by the number and variety of those studied species, when GenoType®, MALDI-TOF MS and rpoβ sequencing methods were compared, it was shown that MALDI-TOF MS was superior respect to GenoType® method to distinguish M. fortuitum complex species. With our results, GenoType® would not be able to distinguish M. fortuitum complex species. However, it would be necessary and interesting to have a greater amount of isolates and species of this complex available in order to be able to verify these results and obtain more solid conclusions. By using MALDI-TOF MS, 4/17 strains of M. fortuitum complex were misidentified. However, it must be pointed out that these four strains showed12 a MALDI-TOF MS score <1.85.
M. mucogenicum was identified only in one sample with MALDI-TOF MS and partial rpoβ sequencing method, while it was identified as Mycobacterium spp. with GenoType®. Han et al.21 described M. mucogenicum as the dominant RGM species responsible for bloodstream and catheter-related infections.
As we have pointed out previously, one limitation of our work would be the total number of different species included in the study. Another limitation of our study were the two strains with a partial rpoβ gene sequencing similarity <98% with respect to the sequences of reference from BLAST. Besides, the phylogenetic analysis found no significant differences between at least two species of Mycobacterium spp.: M. alvei/peregrinum and M. monacense/duvalii. These strains could not be classified correctly due to the short length of the amplified fragment. These strains would be candidates to further study to establish their identification at species level. Interestingly, the 2 strains with partial rpoβ gene sequencing with a similarity <98% showed MALDI-TOF MS scores <1.85 and one of them was identified with phylogenetic analysis. The sequencing of additional regions (complete 16S rRNA, hsp65, sodA, dnaK genes)22–24 would be indicated in these cases. It is known that sequence analysis of DNA isolated from clinical samples has also allowed the discovery of new bacterial pathogens.25
When MALDI-TOF MS score was <1.85, the revision of these MALDI-TOF MS spectra is proposed with the aim of completing the Mycobacterium spp. MALDI-TOF data base.
Another interesting aspect would be the enlargement of the study with slowly growing NTM. However, this would present a greater difficulty due to the intrinsic characteristics of these NTM, as well as the wide diversity of existing species.
In summary, molecular methods are more costly and time-consuming and they may not be available in some clinical microbiology laboratory. Subsequently, our results, which were obtained with a limited number of rapidly growing mycobacteria species, indicate that the MALDI-TOF MS analysis could be a quick identification method in the future. In addition, to identify new species and subspecies of Mycobacterium spp., as well as to identify correctly all clinically relevant Mycobacterium spp., it will be required the sequencing of additional genes of Mycobacterium spp. or whole genome sequencing and the subsequent phylogenetic analysis.
Conflict of interestThe authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.

