


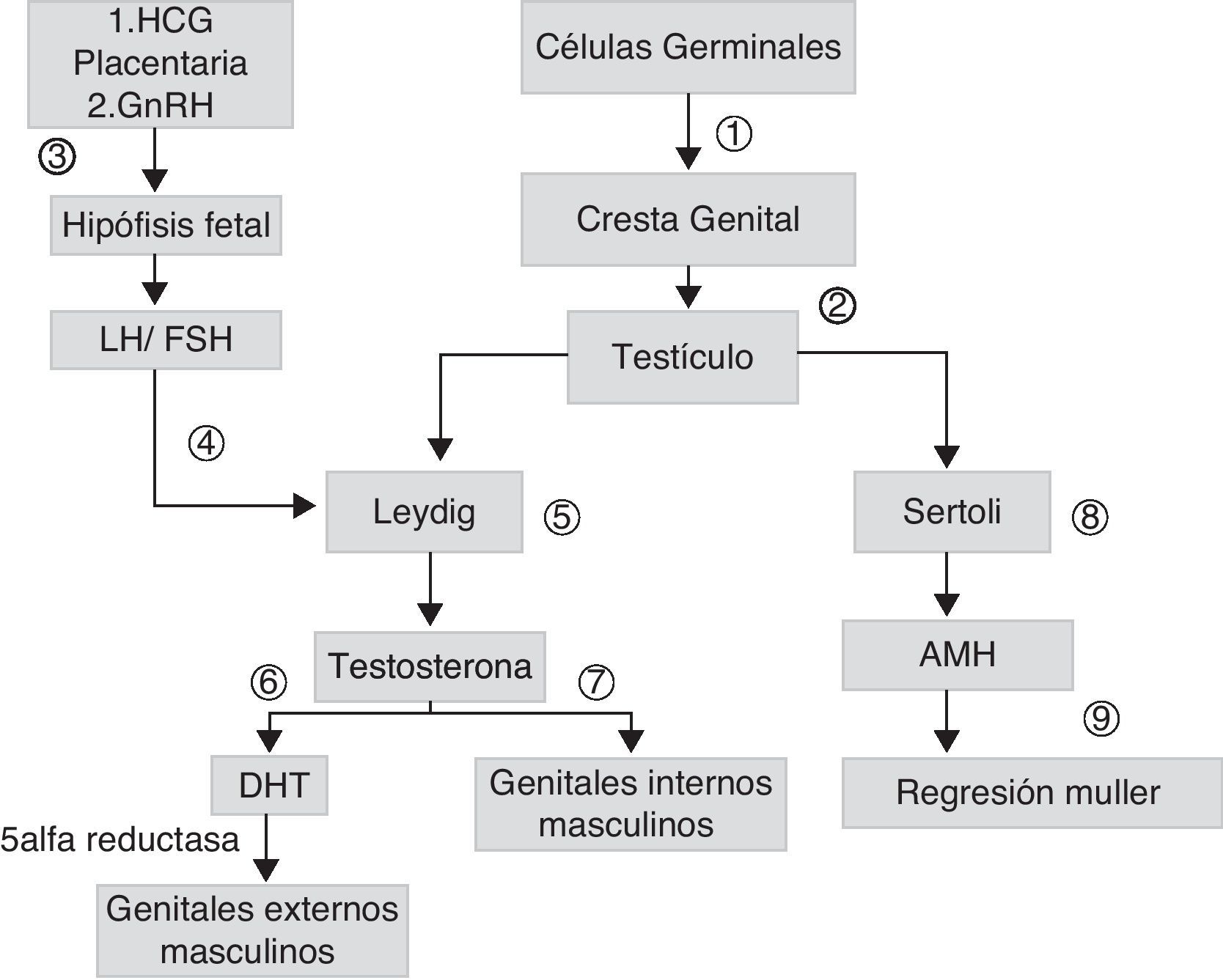
La diferenciación sexual es un proceso que se inicia precozmente durante la embriogénesis. En los varones, el gen SRY, ubicado en el brazo corto del cromosoma Y, codifica para el factor determinante testicular que determina la diferenciación de la gónada hacia testículo. La síntesis de andrógenos testiculares por las células de Leydig se inicia a partir de la octava semana del desarrollo, estimulada inicialmente por la gonadotrofina coriónica (HCG) placentaria y, posteriormente, por la hormona luteinizante (LH). Las gonadotrofinas fetales estimulan la producción a nivel testicular de hormona antimulleriana (AMH), que estimula la regresión de los conductos de Muller en los varones. La testosterona, cuya acción se inicia en la novena semana una vez que el receptor de andrógenos (RA) se encuentra presente, permite estabilizar los conductos de Wolff y su diferenciación hacia epidídimo, vas deferens y vesículas seminales. La conversión de la testosterona en su metabolito más activo, la dehidrotestosterona (DHT) permite el crecimiento del tubérculo genital dando origen al pene y el cierre del rafe ventral, originando la uretra peneana y el rafe escrotal (fig. 1).
, forman los cordones sexuales primitivos dando origen a los testículos (2). Las células de Leydig, son estimuladas por la gonadotrofina coriónica (HCG) placentaria y, posteriormente, por hormona luteinizante (LH) (4), que depende de la hormona liberadora de gonadotrofinas (GnHRH) secretada por el hipotálamo fetal (3), que estimula la producción de gonadotrofinas. Estas estimulan la producción de andrógenos (7) y de hormona antimulleriana (AMH) (8), que permite la regresión de los conductos de Muller (9). La testosterona secretada inicialmente (7), permite el desarrollo de genitales internos masculinos. La conversión de testosterona a dehidrotestosterona (DHT) permite la formación de los genitales externos masculinos (6).")
Esquema de desarrollo sexual embrionario: las células germinales migran hacia las crestas genitales (1), forman los cordones sexuales primitivos dando origen a los testículos (2). Las células de Leydig, son estimuladas por la gonadotrofina coriónica (HCG) placentaria y, posteriormente, por hormona luteinizante (LH) (4), que depende de la hormona liberadora de gonadotrofinas (GnHRH) secretada por el hipotálamo fetal (3), que estimula la producción de gonadotrofinas. Estas estimulan la producción de andrógenos (7) y de hormona antimulleriana (AMH) (8), que permite la regresión de los conductos de Muller (9). La testosterona secretada inicialmente (7), permite el desarrollo de genitales internos masculinos. La conversión de testosterona a dehidrotestosterona (DHT) permite la formación de los genitales externos masculinos (6).
El síndrome de insensibilidad completa a andrógenos (SICA) es un desorden del desarrollo sexual XY en que existe una pérdida de función del RA. Se presenta en individuos con cariotipo 46 XY con genitales externos de fenotipo femenino.
Presentamos el caso de un recién nacido a término producto de un embarazo fisiológico, primera hija de padres sanos no consanguíneos. El peso de nacimiento fue 3.420 g y la longitud de nacimiento 50cm, con Apgar 9-10. Al examen físico presentaba genitales externos femeninos normales y 2 masas de 1cm en ambos conductos inguinales, por lo que se sospechó una hernia inguinal. La ecografía abdominal demostró la presencia de un proceso peritoneo-vaginal persistente en ambas regiones inguinales con herniación del contenido abdominal y 2 formaciones ovoídeas homogéneas de un centímetro de diámetro con flujo al doppler en su interior, sin folículos sugerentes de gónadas. A los 2 días de vida presentaba LH y hormona folículo estimulante (FSH) menores a 0,1 IU/L (valores de referencia (VR): 0,02–7,0 mIU/l), testosterona de 1,45 nmol/L (VR: 0,42–0,72 nmol/L) y estradiol <73,4pmol/L (esperable niveles marcadamente elevados).
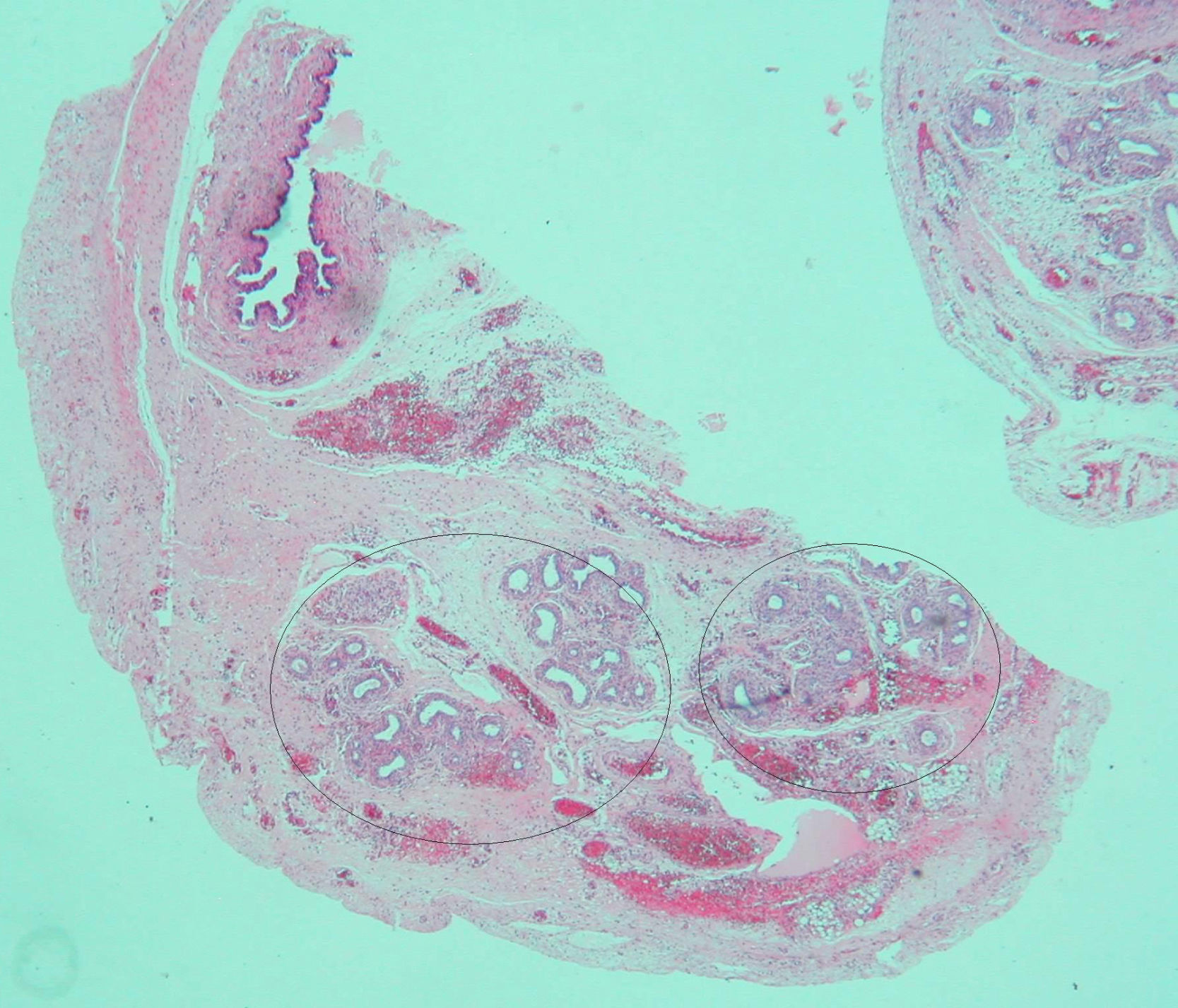
Frente a la sospecha de gónadas en canal inguinal, la paciente fue sometida a cirugía al tercer día de vida. Una vaginoscopia intraoperatoria no mostró cuello uterino. La biopsia gonadal mostró un fragmento de parénquima testicular constituido por cordones sólidos con células de Sertoli y gonias, sin identificar células de Leydig. La biopsia de gónada izquierda mostraba un elevado número de gonias, hidátide quística y numerosas estructuras tubulares compatibles con epidídimo. En la vecindad, presentaba un conducto compatible con resto mulleriano (fig. 2). Se restituyeron ambos testículos en el abdomen.
 que podrían corresponder a remanentes mullerianos.")
El estudio del cariotipo fue 46 XY. El estudio molecular del gen del RA halló un cambio de base en el exón 5, que sustituía una arginina por glutamina en el codón 752 (R752X), mutación comunicada previamente en la literatura en un paciente con SICA1, lo que confirmó el diagnóstico. El estudio molecular de la madre fue positivo para la misma mutación. Esta no presentaba alteraciones fenotípicas.
El SICA es una enfermedad autosómica recesiva ligada al cromosoma X, causada por una mutación en el RA que puede ser heredada (70%) o producirse de novo (30%). El gen se ubica en la región cromosómica Xq11- 12, y está compuesto por 8 exones que codifican para una proteína de 919 aminoácidos. Tiene 4 dominios funcionales: NTD (N- terminal transactivation domain) codificado en el exón 1, DBD (DNA binding domain) codificado en los exones 2 y 3, una región bisagra, y LBD (Ligand binding domain) codificado por los exones 4 a 82. El RA se encuentra en el citoplasma celular unido a proteínas, y al unirse el ligando se trasloca al núcleo donde se une al DNA. Una misma mutación puede producir diferentes fenotipos en la misma familia afectada3.
La prevalencia se estima entre 2 a 5 en 100.0004. Se presenta clínicamente en individuos con cariotipo 46 XY y genitales externos femeninos normales, una vagina con fondo ciego y testes intra-abdominales. Las células de Leydig producen testosterona en cantidad adecuada para un varón de esa edad, la cual es transformada a dehidrotestosterona (DHT) a través de la 5-alfareductasa en forma normal, pero al presentar un RA no funcionante el efecto de la DHT es prácticamente nulo. La producción de AMH por las células de Sertoli y la síntesis de su receptor AMHR2 no estarían afectadas por la alteración del RA. Esto permitiría una acción normal de la AMH durante el período embrionario, explicando la regresión de estructuras mullerianas (tercio superior de la vagina, útero y ovarios) en pacientes con SICA.
El SICA debe sospecharse en 2 instancias: en un recién nacido fenotípicamente femenino con hernia inguinal bilateral, puesto que estas son poco frecuentes en mujeres. Entre 1 y 2% de estas lesiones corresponderían a SICA5. La segunda forma de presentación es en la adolescente con desarrollo mamario adecuado y vello púbico y/o axilar escaso o ausente, con amenorrea primaria, en que se demuestra la ausencia útero y ovarios.
La LH y la testosterona durante los 3 primeros meses de vida son normales o en el límite superior. En pacientes que conservan las gónadas, la reactivación del eje hipotálamo-hipofisiario durante la pubertad induce la actividad de la célula de Leydig, elevando los andrógenos intratesticulares y permitiendo la detención de la proliferación de las células de Sertoli. Por insensibilidad del RA en el núcleo de estas, se mantiene la producción de AMH6. La testosterona y la LH se mantienen elevadas por resistencia androgénica hipotálamo-hipofisiaria. La testosterona se aromatiza a estrógenos en forma periférica, lo que explica el desarrollo mamario femenino normal observado en estos casos7. Parece ser seguro mantener las gónadas in situ hasta la pubertad, puesto que el riesgo de gonadoblastoma u otros tumores aumentaría después de este momento8.
La presencia de restos mullerianos ha sido comunicada en forma esporádica y no tiene consecuencias clínicas. Se han sugerido diversas hipótesis respecto a su persistencia, como 1 la presencia de defectos en la secreción o función de la AMH 2, la ausencia de respuesta de los tejidos mullerianos a la acción de la AMH, por la presencia de altos niveles de estrógenos (secundarios a la conversión periférica de testosterona), o 3 un descenso testicular precoz, que ubica a las estructuras mullerianas fuera del rango de la acción de la AMH durante la embriogénesis9. Estas hipótesis fueron evaluadas en un feto de 20 semanas con diagnóstico prenatal de SICA y presencia de remanentes mullerianos. Se observaron 3 fenómenos, primero, un aumento de la expresión de AMH por las células de Sertoli, segundo, una menor expresión de células mesenquimáticas peritubulares positivas para el receptor de AMH (AMHR2) y, finalmente, hiperplasia de células de Leydig. Estos hallazgos sugieren una nueva hipótesis respecto a la acción de la AMH durante el período embrionario, basada en una resistencia prenatal a AMH10.
En conclusión, la presencia de estructuras mullerianas menores en los casos de SICA puede ser más frecuente de lo hasta ahora comunicado, no debe hacer dudar del diagnóstico y no modifica la evolución ni el tratamiento.
Agradecemos al Dr. Alejandro Martínez-Aguayo por la revisión crítica del manuscrito.
