





La acromegalia y el gigantismo se deben a la producción excesiva de GH, generalmente por un adenoma hipofisario. Es una enfermedad poco frecuente.
El diagnóstico se realiza ante un paciente con un cuadro clínico sugerente con la demostración de concentraciones de GH e IGF-I elevadas. Tras la confirmación bioquímica del exceso de GH debe realizarse una RM del área hipotálamo-hipofisaria a fin de confirmar el origen del exceso de GH.
El tratamiento de elección es el quirúrgico del adenoma hipofisario mediante cirugía transesfenoidal, si bien en los últimos años los avances en cuanto a la aparición de nuevos fármacos han modificado la secuencia terapéutica. El tratamiento médico con análogos de somatostatina puede estar indicado como procedimiento primario en pacientes no subsidiarios de curación tras cirugía, o en aquellos casos en que esta esté contraindicada. El antagonista del receptor de GH debe utilizarse en pacientes no controlados tras cirugía que no responden de forma adecuada a análogos de somatostatina.
La radioterapia estaría indicada en aquellos casos no controlados tras tratamiento quirúrgico y médico o en aquellos pacientes con grandes restos tumorales tras el tratamiento quirúrgico.
La acromegalia y el gigantismo se deben a la producción excesiva de GH, generalmente por un adenoma hipofisario. El diagnóstico se ve precedido de forma invariable por unos 10 años de enfermedad desconocida1.
Los adenomas hipofisarios son responsables de aproximadamente el 15% de los tumores intracraneales. La incidencia de la acromegalia es de aproximadamente 5 casos por millón por año, y la prevalencia, de 60 casos por millón.
Los adenomas somatotropos son de origen monoclonal y se desarrollan a partir de cambios genéticos. GHRH y somatostatina hipotalámicas y paracrinas, al igual que factores de crecimiento, facilitan la expansión de las células somatotropas tumorales1.
Más del 90% de los pacientes con acromegalia presentan un adenoma hipofisario benigno monoclonal, rodeado de tejido hipofisario no hiperplásico. Los adenomas densamente granulados crecen lentamente y se presentan en pacientes mayores de 50 años. Los adenomas escasamente granulados crecen más rápidamente y se presentan en pacientes más jóvenes. Alrededor del 25% de los adenomas secretores de GH cosecretan prolactina; estos incluyen adenomas dimorfos con células de GH y prolactina, adenomas monomorfos mamosomatotropos y adenomas de células acidófilas más primitivas. La inmunorreactividad mixta multicelular o unicelular es frecuente, especialmente para la subunidad alfa de las hormonas glucoproteicas. Raramente existe secreción de otras hormonas con repercusión clínica.
Los adenomas hipofisarios que se desarrollan en los niños antes de completar el crecimiento provocan gigantismo. El gigantismo hipofisario es muy raro, en una amplia serie de 2.367 niños y adolescentes con adenomas hipofisarios, solo el 0,6% presentaban gigantismo2.
Más del 70% de los adenomas somatotropos son macroadenomas al diagnóstico, pero el carcinoma de la célula somatotropa es excepcional. La secreción ectópica de GH es igualmente excepcional. Los síndromes familiares de acromegalia son muy raros. La producción excesiva de GHRH de origen central hipotalámico (generalmente gangliocitomas) o periférico puede dar lugar a hiperplasia de las células somatotropas y acromegalia.
ClínicaLas manifestaciones clínicas de la acromegalia incluyen: los cambios faciales, el crecimiento de las partes acras, prognatismo, hiperhidrosis, cefalea, parestesias, disfunción sexual, hipertensión arterial, bocio, crecimiento de partes blandas, artralgias, síntomas de hiperglucemia, osteoartropatía, miocardiopatía, insuficiencia cardíaca, apnea del sueño e insuficiencia respiratoria. También se presentan síntomas derivados de las manifestaciones locales del tumor, como las alteraciones visuales. Es frecuente la presencia de visceromegalias, en forma de bocio, hepatomegalia, esplenomegalia y macroglosia. Las manifestaciones sutiles de crecimiento de partes acras, crecimiento óseo y aumento de partes blandas se desarrollan de forma inexorable a lo largo de años. Son especialmente características la prominencia frontal, el prognatismo, el engrosamiento cutáneo y el aumento del tamaño de los zapatos y de los anillos (GradoC) (tabla 1).
Manifestaciones clínicas y analíticas de la acromegalia
| Efecto compresivo del adenoma hipofisario |
| Cefalea |
| Defectos campimétricos |
| Hiperprolactinemia |
| Compresión del tallo hipofisario |
| Hipopituitarismo |
| Hipotiroidismo, hipogonadismo, hipocortisolismo |
| Efectos sistémicos del exceso de GH/IGF-I |
| Visceromegalias |
| Cambios cutáneos y de partes blandas |
| Engrosamiento de partes acras |
| Engrosamiento cutáneo e hipertrofia de tejidos blandos |
| Hiperhidrosis |
| Papiloma cutáneo y acantosis nigricans |
| Manifestaciones cardiovasculares |
| Hipertrofia biventricular o septal asimétrica |
| Insuficiencia cardíaca congestiva |
| Enfermedad coronaria |
| Arritmias |
| Hipertensión |
| Miocardiopatía |
| Manifestaciones metabólicas |
| Intolerancia a la glucosa |
| Diabetes mellitus |
| Resistencia a la insulina |
| Manifestaciones respiratorias |
| Macroglosia |
| Maloclusión de la mandíbula |
| Obstrucción de la vía aérea superior |
| Alteraciones del sueño |
| Apnea del sueño (central y obstructiva) |
| Alteración en la ventilación |
| Manifestaciones óseas y articulares |
| Aumento del grosor del cartílago articular |
| Artralgias y artritis |
| Síndrome del túnel carpiano |
| Osteopenia |
| Otras manifestaciones endocrinológicas |
| Bocio |
| Hipercalciuria |
| Galactorrea |
| Disminución de la libido, disfunción eréctil |
| Irregularidades menstruales |
Modificada a partir de: Cordero RA, Barkan AL. Diagnosis of acromegaly. Endocr Metab Disord. 2008;9:13–19.
La acromegalia se desarrolla de forma insidiosa, resultando en un retraso en el diagnóstico de unos 8 a10 años. Las manifestaciones clínicas en cada paciente dependen de las concentraciones de GH e IGF-I, la edad, el tamaño del tumor y el retraso en el diagnóstico3,4.
DiagnósticoLa mayoría de los pacientes se presentan con clínica evidente.
El diagnóstico de acromegalia requiere la demostración de concentraciones elevadas de GH e IGF-I. En la acromegalia las concentraciones de GH están tónicamente elevadas, por tanto, un valor al azar de GH inferior a 0,04μg/l excluye su diagnóstico, pero un valor al azar elevado no implica su presencia.
El diagnóstico bioquímico se realiza con la determinación de las concentraciones de IGF-I en ayunas, y de GH antes y después de la sobrecarga oral de glucosa (SOG) con 75g. El diagnóstico se complica en ocasiones por las variaciones fisiológicas de la GH y por la ausencia de uniformidad de los ensayos para su determinación. Los nuevos inmunoanálisis basados en anticuerpos monoclonales son más sensibles, pero presentan importantes problemas de reproducibilidad5.
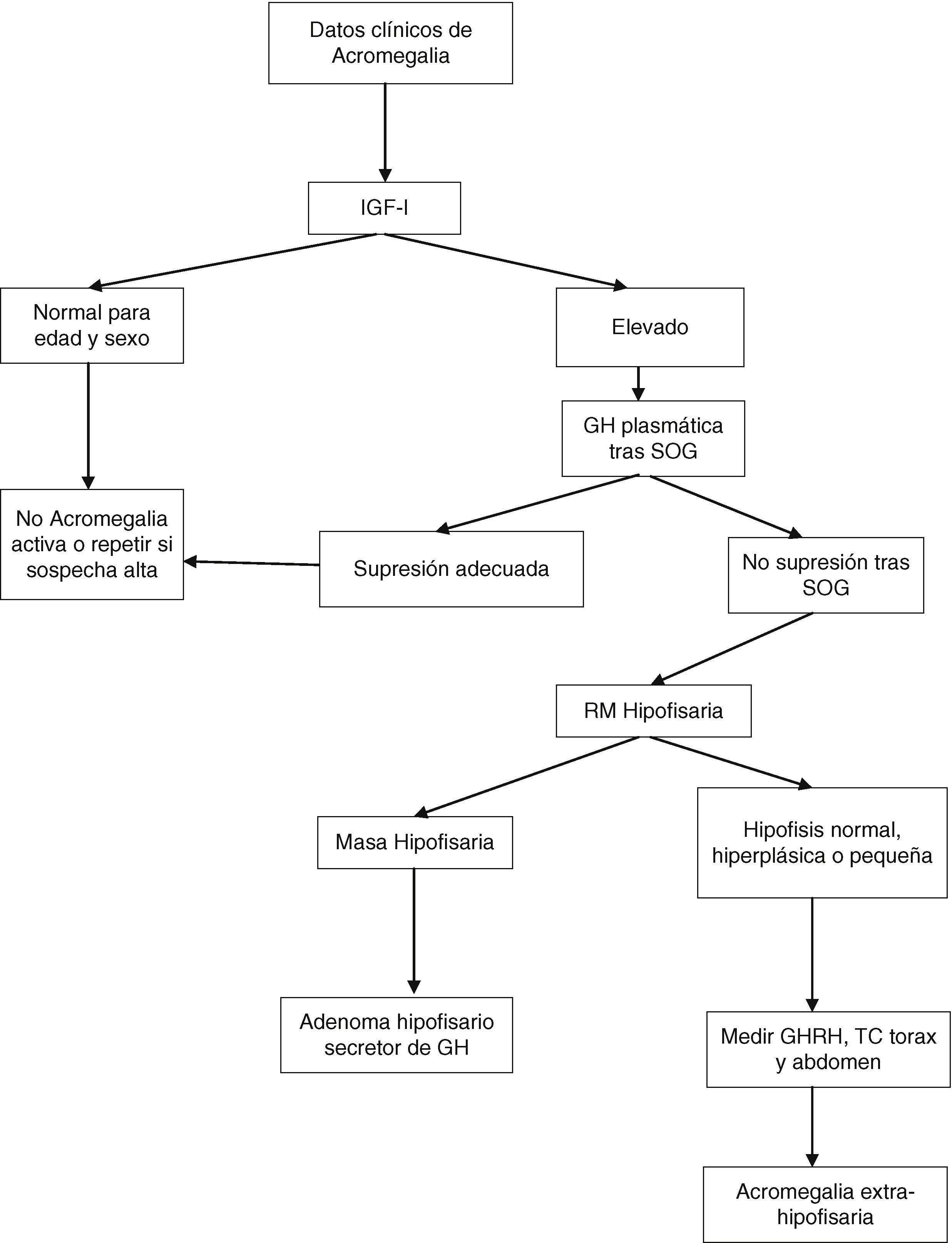
Tras la SOG, los valores nadir de GH por debajo de 1μg/l, con la mayoría de los métodos, descartan la acromegalia. Sin embargo, si se emplean los métodos ultrasensibles de determinación de GH pueden existir pacientes con acromegalia que supriman por debajo de 1μg/l; con alguno de estos métodos el criterio de supresión es una concentración circulante igual o menor de 0,4μg/l (dependiendo del ensayo utilizado) según el último consenso sobre curación de la acromegalia, o incluso de 0,3μg/l según otros autores1,6,7. La GH puede no suprimirse en presencia de enfermedad hepática, renal, diabetes mellitus mal controlada, malnutrición, anorexia, embarazo o tratamiento estrogénico, o en la adolescencia tardía (fig. 1).

Algoritmo diagnóstico de la acromegalia. SOG: sobrecarga oral de glucosa.
Modificado a partir de: Cordero RA, Barkan AL. Diagnosis of acromegaly. Endocr Metab Disord. 2008;9:13-19, y Giustina et al.6
En diferentes situaciones, por tanto, IGF-I sirve como un biomarcador de la actividad de la acromegalia. Las concentraciones de IGF-I son relativamente estables, se correlacionan con los datos clínicos de acromegalia y con las concentraciones plasmáticas elevadas de GH. El nivel plasmático de IGF-I no se eleva más cuando las concentraciones de GH son mayores de 20μg/l, y elevaciones sutiles de GH no siempre elevan el nivel circulante de IGF-I. La valoración precisa de IGF-I requiere controles ajustados por edad, pues sus concentraciones disminuyen un 14% con cada década. En el seguimiento de la enfermedad la determinación de GH e IGF-I circulantes es complementaria.
La resonancia magnética (RM) hipofisaria con administración de gadolinio es la mejor técnica de imagen para localizar el origen del exceso de GH. Esta técnica permite ver y localizar, en relación con las estructuras vecinas, los adenomas mayores de 2mm de diámetro. En el momento del diagnóstico, más de un 75% presentan un macroadenoma (superior a 10mm), que crece hacia el seno cavernoso o supraselar. En los casos raros, en que se sospecha un origen no hipofisario, debe realizarse tomografía computarizada o RM torácica y abdominal o una gammagrafía con octreoscan.
TratamientoLos objetivos del tratamiento de la acromegalia son:
- 1.
Control del crecimiento tumoral.
- 2.
Normalizar las concentraciones elevadas de IGF-I y GH.
- 3.
Control de los síntomas, mejora de la calidad de vida y control de las comorbilidades.
- 4.
Prevención de la mortalidad prematura (GradoB).
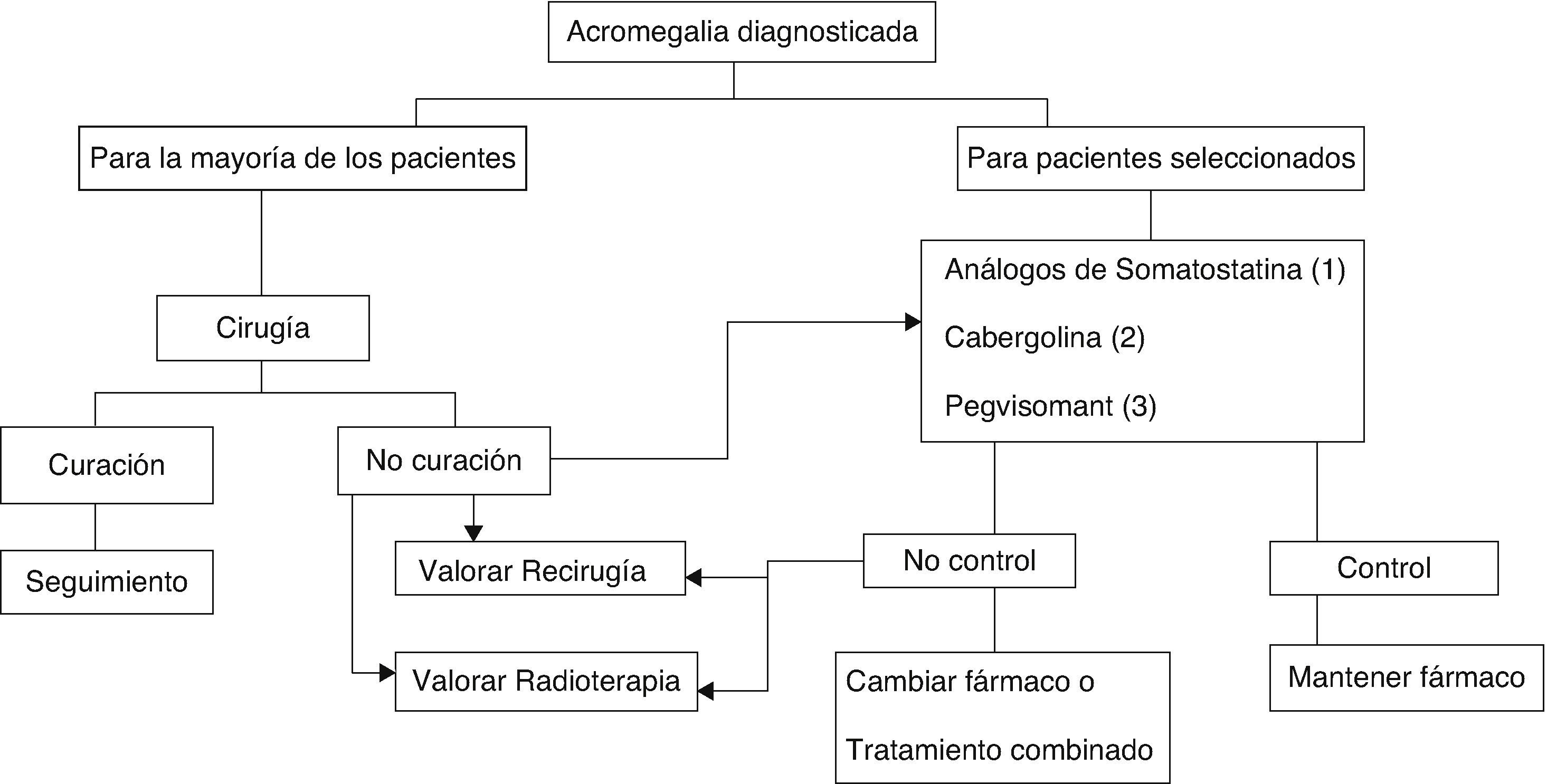
En la actualidad existen 3 modalidades terapéuticas: tratamiento quirúrgico, tratamiento médico y radioterapia6,8,9 (fig. 2).
 De elección, si no existen contraindicaciones o intolerancia. Tratamiento primario en los que por cualquier razón no se intervengan. (2) A valorar: muy ancianos, discreta elevación de GH e IGF-I, tumores mixtos productores de GH y prolactina. (3) No admitido en ficha técnica como primera opción farmacológica. A valorar en sujetos con clínica muy intensa o IGF-I muy elevada.")
Algoritmo terapéutico de la acromegalia.
(1) De elección, si no existen contraindicaciones o intolerancia. Tratamiento primario en los que por cualquier razón no se intervengan.
(2) A valorar: muy ancianos, discreta elevación de GH e IGF-I, tumores mixtos productores de GH y prolactina.
(3) No admitido en ficha técnica como primera opción farmacológica. A valorar en sujetos con clínica muy intensa o IGF-I muy elevada.
El tratamiento quirúrgico de la acromegalia sigue siendo el de elección en la mayoría de los pacientes (GradoB), si bien los avances ocurridos en el tratamiento médico en los últimos años han modificado la secuencia terapéutica6,8,9.
La radioterapia ocupa en la actualidad el último escalón en el esquema terapéutico, quedando reservada para pacientes no controlados tras tratamiento médico o quirúrgico inicial, y en caso de macroadenomas invasivos no controlados (GradoC)6,9.
Es importante que el paciente sea atendido por un equipo multidisciplinar en el que estén implicados endocrinólogos, neurocirujanos y radioterapeutas expertos, a fin de aconsejar el tratamiento más adecuado en cada caso (GradoC)6,8.
Tratamiento quirúrgicoEs el tratamiento de primera línea en la mayoría de los pacientes. Es el procedimiento de elección en microadenomas, macroadenomas con síntomas compresivos y macroadenomas subsidiarios de curación quirúrgica (GradoA). También puede ser el tratamiento de elección en macroadenomas no subsidiarios de curación, a fin de disminuir el tamaño tumoral y facilitar la respuesta al tratamiento complementario (GradoB).
En una reciente encuesta publicada sobre manejo de acromegalia, el tratamiento quirúrgico es el de elección en la mayoría de los pacientes; así, es el tratamiento elegido por los encuestados de Europa y EE. UU. en el 90 y 94% de los microadenomas y en el 92 y 94% de los macroadenomas con compromiso visual, respectivamente. El porcentaje desciende en casos de macroadenomas sin compresión de estructuras vecinas, y en aquellos no subsidiarios de curación tras cirugía10.
Ventajas de la cirugíaLa disminución del tamaño tumoral o la extirpación completa del mismo pueden inducir la curación, produciendo un descenso rápido de los niveles hormonales que detiene la progresión de la enfermedad y mejora las comorbilidades.
En macroadenomas invasivos, no subsidiarios de curación, la extirpación de masa tumoral produce descompresión rápida de las estructuras adyacentes y del tejido hipofisario normal, preservando la función hipofisaria, facilitando, además, la respuesta al tratamiento médico o radioterápico posterior.
Permite obtener tejido tumoral para estudio inmunohistoquímico, ultraestructural y molecular, lo que ayuda a seleccionar el mejor tratamiento médico posterior.
Preparación del paciente antes de la cirugíaDeberá realizarse una historia clínica detallada, incluyendo estudios analíticos básicos y una valoración hormonal hipofisaria sobre todo en macroadenomas, a fin de confirmar la necesidad de sustitución hormonal del eje adrenal o tiroideo previa a la cirugía (GradoC). También debe evaluarse el riesgo cardiovascular y la presencia de comorbilidades que deban ser tratadas antes de la cirugía9.
Tratamiento preoperatorio con análogos de somatostatinaAlgunos estudios muestran que el tratamiento con análogos de somatostatina (AASS) previo a la cirugía consigue tasas mayores de remisión que en los pacientes no tratados (GradoC); sin embargo, otros estudios no confirman estos hallazgos. En un estudio multicéntrico noruego el pretratamiento con octreótido durante 6 meses consigue tasas de curación (definida como IGF-I normal) en el 50% de los pacientes con macroadenoma, frente al 16% de los pacientes no tratados. No obstante, cuando se añade como criterio de curación la supresión de GH tras glucosa inferior a 1μg/l la diferencia entre los 2 grupos no fue significativa11. En otro estudio de 98 pacientes con macroadenoma tratados con lanreótida durante 4 meses la tasa de remisión tras la cirugía fue del 49% en el grupo tratado frente al 18% en el grupo no tratado. La diferencia entre los 2 grupos permaneció significativa cuando se añade la frenación de GH tras glucosa inferior a 1μg/l12.
En algunos estudios el tratamiento previo con AASS disminuye la incidencia de complicaciones o la estancia hospitalaria, sin embargo, otros estudios no confirman estos hallazgos.
Los AASS mejoran la función cardíaca disminuyendo la hipertrofia ventricular y la incidencia de arritmias, lo que, junto al beneficio sobre el edema tisular de las vías respiratorias que facilita la intubación, los sitúan como una opción de tratamiento conveniente antes de la cirugía (GradoC)13–15.
En la encuesta anteriormente referida el 75% de los encuestados utiliza AASS previamente a la cirugía, el 35% para reducir los riesgos de la anestesia, el 24% para conseguir control bioquímico previo y en el 10% por preferencia del paciente. Aunque el tratamiento médico puede estar indicado, son necesarias nuevas evidencias antes de recomendarlo de forma sistemática en todos los pacientes acromegálicos que van a ser sometidos a cirugía10.
El éxito de la cirugía es dependiente de la experiencia del equipo neuroquirúrgico16(GradoA). El rango de control se correlaciona con la experiencia del neurocirujano; la realización de al menos 50 intervenciones quirúrgicas al año es necesaria para considerar a un neurocirujano como experto.
Técnica quirúrgicaLa cirugía transesfenoidal es la técnica quirúrgica de elección; la craneotomía está raramente indicada en la actualidad en pacientes con acromegalia.
Las nuevas técnicas quirúrgicas endoscópicas, la neuronavegación, el uso de RM intraoperatoria o la determinación de concentraciones intraoperatorias de GH pueden mejorar el seguimiento operatorio, la satisfacción del paciente y disminuir la tasa de complicaciones. Los resultados iniciales son prometedores, pero limitados, por lo que se precisan nuevos estudios para confirmarlos. Son técnicas caras que alargan la duración de la intervención. Su utilización es dependiente de la decisión y experiencia del equipo neuroquirúrgico17.
ResultadosLa eficacia del tratamiento quirúrgico se correlaciona de forma inversa al:
- 1.
Tamaño tumoral.
- 2.
Concentraciones preoperatorias de GH e IGF-I (GradoC).
El tratamiento quirúrgico del microadenoma consigue tasas de curación de entre el 75-95% de los pacientes (GradoB); la tasa de curación en macroadenomas no invasivos es menor, consiguiendo normalización de IGF-I en el 40-68% de los casos10,11. La influencia del tamaño tumoral sobre el éxito quirúrgico es objeto de controversia, pero en general se admite que tumores mayores de 2cm se asocian a un menor número de curaciones (GradoB). En los macroadenomas invasores el porcentaje de curación es escaso. Por otra parte, concentraciones de GH superiores a 30μg/l se asocian con invasión de seno cavernoso y bajo índice de curación quirúrgica (20-50%)18-20(GradoB).
Datos del Registro Español de Acromegalia muestran que el 81,2% de los pacientes fueron tratados con cirugía; el porcentaje de curación fue del 40,3% (definida como GH inferior a 2μg/l tras SOG, IGF-I normal, o ambos)21.
La reintervención podría plantearse en pacientes con restos tumorales visibles en la RM, en los cuales no se consigue curación tras la primera cirugía3.
Manejo perioperatorioTras la cirugía deben monitorizarse diuresis y electrolitos por si el paciente desarrolla diabetes insípida, que puede presentarse en el 10-20% de los pacientes; la mayoría de las veces es transitoria, si bien puede ser permanente en el 2-7% y requerir tratamiento con desmopresina a largo plazo22.
La hiponatremia relacionada con el síndrome de secreción inadecuada de hormona antidiurética se presenta entre los días 5 y 14 después de la cirugía, y afecta al 5-10% de los pacientes. En la mayoría de las ocasiones es leve y solo requiere restricción hídrica, pero en ocasiones es más grave y puede requerir la administración de solución salina hipertónica intravenosa22.
Debe monitorizarse la función adrenal en el postoperatorio inmediato por si fuese necesario el tratamiento sustitutivo glucocorticoideo (GradoC).
Seguimiento postoperatorioUn descenso de GH precoz a las 24-48h (inferior a 2μg/l) se asocia con remisión a largo plazo, si bien se recomienda la medida de IGF-I entre las semanas 9 y 12 tras la cirugía. La normalización de IGF-I se asocia con remisión quirúrgica. Si la IGF-I persistiese elevada a las 12 semanas debe repetirse en otras 12 semanas por si se produce remisión tardía, antes de iniciar tratamiento complementario.
La SOG con 75 g para frenación de GH (con determinaciones cada 30min) se debe realizar a partir de los 3 meses, frenación de GH inferior a 1μg/l se asocia con alta probabilidad de remisión quirúrgica (GradoC), la frenación de GH inferior a 0,4μg/l incrementa la sensibilidad del test6,20.
La RM postoperatoria debe realizarse también a partir del tercer mes tras la cirugía a fin de evaluar si existe tumor residual.
La función hipofisaria debe ser evaluada entre las semanas 6 y 12 tras la cirugía a fin de valorar la necesidad de tratamiento hormonal sustitutivo (GradoC).
Estudio histológicoEs importante la obtención de una muestra por el neurocirujano para estudio histológico. El índice de proliferación Ki67 puede indicar mayor o menor agresividad y orientar a la terapia posterior. Tumores con inmunohistoquímica positiva para GH sin manifestaciones clínicas de exceso de GH son denominados clínicamente silentes, pero pueden tener un comportamiento agresivo y ser subsidiarios de tratamiento con AASS.
Tumores con positividad a prolactina pueden responder mejor a agonistas dopaminérgicos. Los tumores escasamente granulados responden peor al tratamiento con AASS que los densamente granulados. Por último, el estudio molecular de los distintos subtipos de receptores de somatostatina puede orientar hacia un tratamiento posterior más eficaz9.
Tratamiento médicoEl tratamiento médico de la acromegalia puede indicarse:
- 1.
Como tratamiento primario en pacientes que tienen riesgo quirúrgico importante, en los casos en que exista baja probabilidad de curación porque el tumor tenga una extensión extraselar, sin compresión quiasmática, y en aquellos pacientes que no quieren ser operados y optan por el tratamiento médico (GradoC).
- 2.
Como tratamiento complementario, tras fracaso de la cirugía o en el intervalo de tiempo hasta que la radioterapia sea eficaz (GradoB).
- 3.
Podría también indicarse como pretratamiento de la cirugía con la finalidad de mejorar las condiciones anestésicas del paciente o los resultados de la propia cirugía, o en los casos en que la cirugía se demore.
En el momento actual existen 3 grupos de fármacos posibles a utilizar: AASS, agonistas dopaminérgicos y antagonistas periféricos de la GH (GradoA).
Análogos de somatostatinaMecanismo de acciónLos AASS, como la propia somatostatina nativa, ejercen una acción inhibitoria sobre la secreción de GH. Los AASS actualmente disponibles, octreótido y lanreótida, actuarían fundamentalmente a través de los receptores subtipo 2 y en menor medida sobre el subtipo 5, por lo que la respuesta pudiera estar condicionada a la mayor o menor presencia de este tipo de receptores en el tumor (GradoC)23. Se encuentra en desarrollo un nuevo AASS, el SOM-230 o pasireótido, con una mayor potencia de acción a través de receptores subtipo 5, y con un espectro más amplio sobre subtipos 2, 3 y 1, aunque con un peor comportamiento sobre el metabolismo hidrocarbonado24,25. La acción de los AASS está mediada principalmente a través de la subunidad Gα, inhibiendo la adenil-ciclasa y reduciendo la generación de AMP-cíclico. Además, regulan la actividad de tirosinfosfatasa y los canales de calcio y potasio.
Presentaciones y pautas de dosificaciónInicialmente se utilizó octreótido (Sandostatin®) soluble, que debe administrarse 3 o 4 veces al día, 100μg/6 u 8h hasta una dosis máxima de 1.500μg/día. Las ventajas del octreótido soluble son la mayor rapidez de acción, la autoadministración subcutánea y su menor precio, aunque los pacientes prefieren las más recientes presentaciones de vida media larga que requieren administración intramuscular o subcutánea profunda (lanreótida 30mg cada 10 o 14 días) o, mejor, las formas vehiculadas en microesferas de octreótido LAR (Sandostatin® 10, 20 y 30mg) y lanreótida autogel (Somatulina autogel® 60, 90 y 120mg), cuya frecuencia media de administración es cada 28 días. La dosis debe ser individualizada según la respuesta terapéutica. En casos de respuesta eficaz en tratamientos crónicos la dosis puede espaciarse hasta los 42 o 56 días.
La ficha técnica de octreótido recomienda, antes de iniciar tratamiento con la forma LAR, utilizar la forma soluble subcutánea, 100μg/8h durante 2 semanas para valorar la tolerancia, aunque esta también puede establecerse con una o dos dosis (GradoC). La utilidad en la realización de un test agudo como predictor de respuesta futura se cuestiona26,27(GradoC). Una vez comprobada la tolerancia se puede pasar a las formas depot a dosis media (20mg para octreótido LAR, 90mg para lanreótida autogel) y ajustar posteriormente según respuesta.
Durante el tratamiento con análogos se deben determinar las concentraciones de GH e IGF-I o solo de este último. No es necesario el test de supresión de GH tras SOG ni las determinaciones seriadas de GH (GradoD). Hay que tener en cuenta que con valores muy elevados de GH, las concentraciones de IGF-I alcanzan un nivel máximo, por lo que debe esperarse un retraso en la reducción de IGF-I a medida que vaya descendiendo la concentración de GH.
Efectos clínicosLos AASS han demostrado una eficacia probada sobre el control de las concentraciones plasmáticas de GH, así como de IGF-I (GradoB). Con octreótido soluble se consigue una reducción de GH e IGF-I en el 50 a 70% de los pacientes, con normalización de IGF-I en el 30% de los pacientes en los que la cirugía ha fracasado (GradoB)28.
Como tratamiento primario, los AASS permiten un control bioquímico (IGF-I normal para edad y sexo) hasta en un 70% de los pacientes, y con una buena tolerancia. También se ha observado una mayor respuesta de los AASS cuando se combinan con el tratamiento quirúrgico reductor de masa, lo que podría plantearse para pacientes con enfermedad avanzada (GradoC).
Como tratamiento adyuvante, los AASS de acción prolongada consiguen normalizar la IGF-I en el 67% y un valor normal de GH (inferior a 2,5μg/l o valor inferior a 1μg/l tras SOG) en el 57% de los pacientes, según un metaanálisis de 44 ensayos, con mayor respuesta a los 6 meses de tratamiento (GradoB)29. Octreótido LAR y lanreótida autogel ofrecen un perfil de eficacia similar (GradoB).
Se han descrito diversos predictores de respuesta: los mejores predictores son la edad y el sexo (mejor en sujetos ancianos y mejor en mujeres en edad fértil o en tratamiento con estrógenos), pero sobre todo el tamaño tumoral (los tumores más pequeños y menos invasivos), así como los que presentan concentraciones de GH e IGF-I más bajas pretratamiento. Otros predictores de respuesta que se han comunicado son: la ya referida disminución de GH tras una dosis de octreótido soluble (test agudo), cirugía previa, captación de octreótido marcado (octreoscan) (GradoD), radioterapia previa, presencia del oncogén gsp (mutación en la subunidad del complejo Gsα) (GradoD), señal de imagen hipodensa en T2 en la RM (GradoD), así como las características histopatológicas, ya que responden mejor los tumores densamente granulados (GradoC)30–35.
Además de su efecto antisecretor, los AASS han demostrado por su acción antiproliferativa una eficacia en la reducción del tamaño tumoral (GradoB). En un metaanálisis de 14 estudios se concluye que muestran una reducción significativa en hasta casi un 40% de los pacientes, sin diferencia significativa entre la utilización de análogos de acción rápida o los de efecto prolongado, ni tampoco entre los microadenomas y los macroadenomas (GradoC)36,37.
No obstante, hasta un tercio de los pacientes se muestra resistente a la acción de los AASS (Grado B). Esta resistencia se define por insuficiente respuesta bioquímica, así como por fallo en la reducción tumoral mayor de un 20%, aunque pueden definirse respuestas parciales (GradoC). En muchos casos depende del tiempo de tratamiento y de la dosis que pudiera incrementarse hasta 60mg de octreótido LAR (GradoC)28,38.
Efectos adversos (tabla 2)En general, los AASS son bien tolerados. Los efectos adversos más frecuentes son los gastrointestinales (GradoA): diarrea malabsortiva, dolor abdominal tipo retortijón, flatulencia, náuseas y, menos frecuentemente, estreñimiento.
Efectos secundarios de los fármacos utilizados en el tratamiento de la acromegalia
| Octreótido, lanreótida | Cabergolina | Pegvisomant |
| Dolor y eritema en el lugar de inyecciónDolor abdominalDiarrea malabsortivaFlatulencia, hinchazónNáuseas, vómitosAnorexiaLitiasis biliarIntolerancia a la glucosaDéficit de B12Déficit de vitaminas liposolublesBradicardiaAlopeciaAnafilaxia y alergiasPancreatitis | Náuseas, vómitosHipotensión ortostáticaCongestión nasalDisneaCefaleaVértigoInsomnioFibrosis retroperitonealDisfunción valvular | Elevación de transaminasasDéficit funcional de GHLipohipertrofiaDolor en el lugar de inyecciónCefaleaVértigoNáuseas, vómitosDislipidemiaAumento de pesoSíndrome gripal |
Con el uso prolongado se producen cálculos biliares en un 5 a 20% (GradoB). Los cálculos suelen ser asintomáticos, por lo que puede recomendarse un control ecográfico, cuya frecuencia no se ha establecido. Los cálculos sintomáticos deberán ser tratados mediante cirugía o con fármacos que disuelven las sales biliares29,39,40.
La malabsorción puede alterar la absorción de grasas en algún paciente, así como reducir la de vitamina B12.
Menos frecuentemente pueden ocasionar caída del cabello y bradicardia, sobre todo en aquellos pacientes que ya están tomando algún fármaco bradicardizante (betabloqueantes o antagonistas del calcio)41.
Los AASS producen un deterioro en la tolerancia a la glucosa y podrían ocasionar o empeorar una diabetes mellitus ya presente. En estos casos puede disminuirse la dosis si el control de la enfermedad lo permitiese, sustituir por el antagonista de GH u optimizar el control glucémico con los fármacos hipoglucemiantes42,43.
También reducen la absorción de ciclosporina y retrasan la absorción de cimetidina, aumentan la biodisponibilidad de bromocriptina y pueden ocasionar interacción con otros fármacos que se metabolizan mediante las enzimas del citocromo P450, sobre todo con bajo índice terapéutico.
Se han comunicado algunos casos de pancreatitis, lo que resulta paradójico ya que los AASS se utilizan en el tratamiento de estas44.
Agonistas dopaminérgicosFueron los primeros fármacos específicos utilizados en la acromegalia. Su administración por vía oral y su menor coste los hacen especialmente indicados en casos leves en los que la GH e IGF-I están discretamente elevados.
Mecanismo de acciónActúan a través de receptores D-2 de dopamina disminuyendo la hipersecreción de GH (GradoA). Inicialmente se utilizó la bromocriptina, con alguna acción solo en un 10% de los pacientes, por lo que hoy no se recomienda su uso. La cabergolina, agonista más selectivo, es el único dopaminérgico que tiene algún papel en el tratamiento de la acromegalia45.
Dosis y pautas de administraciónSe utiliza la cabergolina (Dostinex®) a dosis variable, pudiendo iniciar con 1mg/sem, pero, a diferencia del tratamiento del prolactinoma, habitualmente se precisan dosis más elevadas, llegando en algunos casos hasta 7mg/sem para conseguir normalizar la alteración bioquímica.
La monitorización de la respuesta debe hacerse como con los AASS, determinando las concentraciones de GH e IGF-I.
Efectos clínicosLos agonistas dopaminérgicos son menos eficaces que los AASS, por lo que, como se ha dicho anteriormente, estarían más indicados en los casos con una discreta elevación de IGF-I y en los tumores mixtos que producen GH y prolactina, aunque las concentraciones de esta no predicen la respuesta en la acromegalia (GradoC)46.
Un metaanálisis publicado recientemente de 10 estudios concluye que la cabergolina en monoterapia consigue una normalización de IGF-I en un tercio de 170 pacientes analizados, con datos limitados sobre disminución del tamaño tumoral(GradoC)47.
Efectos adversos (tabla 2)La cabergolina se tolera mejor que la bromocriptina (GradoC). No obstante, puede producir intolerancia digestiva (náuseas, vómitos y malestar epigástrico), hipotensión arterial ortostática, congestión nasal, disnea y cefalea como efectos adversos más frecuentes, que se presentan en un 10% de los pacientes48.
Con el uso prolongado de estos agentes y cuando se utilizan dosis elevadas (por encima de 3mg/sem de cabergolina) puede producirse disfunción de las válvulas cardíacas. Este efecto adverso puede presentarse en la enfermedad de Parkinson, donde las dosis que se utilizan son mucho más altas, pero no se ha demostrado en la acromegalia49,50, a pesar de que la dosis puede ser frecuentemente mayor que en el prolactinoma. De todas formas se recomienda un control ecocardiográfico sobre todo en los casos en los que la dosis de cabergolina sea elevada, que puede repetirse cada 3 años si fuese normal.
Antagonista del receptor de la hormona del crecimiento: pegvisomantMecanismo de acciónEl pegvisomant (PEG), único antagonista de GH, es un producto pegilado de 199 aminoácidos, obtenido por recombinación genética a través de Escherichia coli, que se comporta como un antagonista selectivo del receptor de GH. La sustitución de glicina 120 en la tercera cadena α hélix de la molécula de GH interfiere la segunda unión con el receptor. Otras 8 sustituciones en aminoácidos aumentan su capacidad de unión en el primer paso y la pegilación le confiere una menor antigenicidad, aumentando su vida media. No suprime la dimerización y el complejo GHR es internalizado, pero incapaz de producir una señal de transducción intracelular. No disminuye la GH porque no tiene efecto directo sobre el tumor hipofisario51,52.
Dosis y pautas de administraciónEl PEG se presenta en viales para administración subcutánea de 10, 15 y 20mg. La ficha técnica, y así se hizo en los ensayos clínicos, aconseja iniciar con una dosis de 80mg para conseguir concentraciones adecuadas, y proseguir con una dosis que generalmente oscila entre 10 y 20mg/día53,54. Algunos pacientes pueden requerir hasta 40mg/día. Dado su elevado coste, se han utilizado regímenes de administración semanal o 2 veces a la semana, lo que puede ser de utilidad en determinados pacientes (GradoC)55.
Durante el tratamiento con PEG se debe monitorizar el IGF-I para valorar su eficacia (GradoB). No tiene sentido determinar concentraciones de GH, que debe aumentar pero que no tiene valor para la dosificación (GradoB), ya que, además, el resultado puede ser alterado por interferencia en el método analítico.
Efectos clínicosPEG es el fármaco que controla mejor las concentraciones de IGF-I en la acromegalia (GradoA), aunque en el momento actual solo está autorizado su uso en el fracaso de AASS o cuando estos no puedan utilizarse por sus efectos adversos.
Los primeros ensayos clínicos con el fármaco administrado a dosis de 10, 15 o 20mg frente a placebo demostraron una reducción de IGF-I en el 60% a las 12 semanas, con una normalización del mismo de hasta un 89%, y en estudio abierto de 18 meses en un 97% de los pacientes junto a una disminución en los síntomas (edema, artralgias) y signos (tamaño del anillo, etc.) de forma dependiente de dosis (GradoB)53,54.
En el German Pegvisomant Observational Study, con 229 acromegálicos medicados con PEG, pero que habían sido tratados previamente mediante cirugía (90,4%), radioterapia (43,2%) y tratamiento médico (94,3%), se logró una normalización de IGF-I del 70,9% a los 12 meses y del 76,3% a los 24 meses56.
Esta menor eficacia en los estudios observacionales probablemente es debida a que las dosis utilizadas no han sido convenientemente ajustadas, y a que el tratamiento con PEG se ha indicado en casos de resistencia a otros tratamientos.
De forma paralela a la disminución de IGF-I se produce un incremento en las concentraciones de GH, que alcanzan una estabilización a los 6 meses de tratamiento de 12 a 14ng/ml. Ello se debe a la interrupción de la retroalimentación negativa al disminuir la producción de IGF-I.
Algunos factores pueden influir en la respuesta terapéutica, habiéndose señalado que el sexo femenino y el mayor índice de masa corporal precisan una mayor dosis, así como la concentración previa de GH e IGF-I; por el contrario, la radioterapia previa mejoraría la respuesta57.
A diferencia de los AASS, diversos estudios demuestran que el PEG mejora la homeostasis de la glucosa, por lo que podría ser una opción en pacientes diabéticos o con intolerancia a la misma (GradoC)58.
Efectos adversos (tabla 2)Una preocupación que surge en el tratamiento con PEG es la posibilidad de un crecimiento tumoral por la pérdida de la retroalimentación negativa ya comentada, con un efecto Nelson-like, y porque este fármaco no posee acción antiproliferativa.
Se han comunicado unos pocos casos de crecimiento tumoral (3,2% en el estudio German Pegvisomant Observational Study), aunque probablemente pueda relacionarse con la historia natural del tumor, ya que los cambios en el volumen no dependen de la duración del tratamiento y se relacionan con tumores de mayor agresividad o por un efecto rebote tras retirada de los AASS (GradoC)59–61. La radioterapia parece disminuir el potencial de crecimiento (GradoC), que también se ha relacionado con el tiempo de tratamiento con análogos (GradoD)62.
Por todo ello se recomienda seguimiento con RM cada 6 meses en el primer año y posteriormente cada año (GradoC), y sería prudente evitar el tratamiento con PEG en monoterapia en pacientes con grandes macroadenomas o tumores muy próximos al quiasma óptico (GradoC).
En un 2,5% de los pacientes tratados con PEG se produce una elevación de las enzimas hepáticas 3 veces sobre el rango alto de normalidad, con datos de 1.288 pacientes de la base de ACROSTUDY63(GradoB). Esta alteración suele presentarse en los primeros 3 meses de tratamiento y, por lo general, es autolimitada y no se relaciona con la dosis (GradoB), aunque sí con el tratamiento previo con AASS64(GradoD). Se recomienda, por tanto, la monitorización analítica de las pruebas de función hepática de forma periódica y la suspensión del tratamiento en los casos de alteración severa o síntomas y signos de hepatitis (GradoC).
El tratamiento crónico con PEG, por su mecanismo de acción, produce un déficit funcional de GH, por lo que el objetivo de tratamiento debería ser el mantenimiento de IGF-I en el rango alto de normalidad, ajustado por edad y sexo65.
Otros efectos adversos del PEG menos frecuentes son el desarrollo de lipohipertrofia en la zona de inyección, que suele también presentarse en las primeras semanas de tratamiento y que cede al suspenderlo66. Más raramente pueden producirse reacciones alérgicas, dolor y eritema local tras la inyección, mareos, cefalea y síndrome gripal (GradoC)67.
Tratamientos combinadosEn algunos pacientes en los que no se consigue una respuesta óptima clínica o bioquímica podría plantearse la asociación de los fármacos descritos. En general, los tratamientos combinados consiguen una mayor efectividad y pueden hacer disminuir las dosis y, por tanto, la toxicidad, aunque un aspecto importante a considerar sería el coste económico68,69.
La asociación de cabergolina en pacientes no controlados con AASS consiguió una normalización de IGF-I en el 40% en un estudio. La respuesta no se relaciona con hiperprolactinemia ni con inmunohistoquímica positiva a prolactina en el tumor (GradoC)70,71.
Otros estudios recientes valoran la asociación de PEG y cabergolina72,73. En el primero, la adición de PEG a dosis fija de 10mg a 0,5mg/día de cabergolina normalizó IGF-I en un 68%. Al retirar la cabergolina solo el 26% mantenían IGF-I en los límites normales.
La asociación de PEG a AASS constituye una opción atractiva por su distinto y complementario mecanismo de acción. La asociación en la mayoría de los trabajos parece más eficaz en disminuir los niveles de IGF-I que cada fármaco por separado74, aunque en un estudio de pacientes resistentes a AASS la asociación no fue superior a PEG solo75. Además, permite disminuir la frecuencia de la inyección de PEG en una o 2 dosis semanales76.
La combinación tiene la ventaja de que los AASS pueden disminuir el tamaño tumoral y el PEG puede mejorar el efecto deletéreo de estos sobre la homeostasis de la glucosa (GradoC). Como desventaja puede esperarse una mayor toxicidad hepática con elevación de enzimas, que en el estudio referido ocurrió en un 11-15% de los casos, con un riesgo mayor en pacientes diabéticos74.
Son necesarios, por tanto, estudios más amplios que puedan clarificar en el futuro la terapia de combinación.
A continuación proponemos un algoritmo de tratamiento basado fundamentalmente en las características particulares de cada paciente. Con las distintas estrategias de tratamiento, en el momento actual hasta un 95% de los pacientes puede alcanzar una remisión completa77 (fig. 2).
RadioterapiaLa radioterapia es considerada como tratamiento de tercera línea en pacientes no controlados tras cirugía y en aquellos no respondedores a tratamiento médico (GradoC), si bien en algunas ocasiones puede ser considerado de segunda línea8,9,78.
La radioterapia debe ser considerada en pacientes no controlados tras cirugía con el objetivo de acortar la duración del tratamiento médico (GradoC).
Tipos de radioterapiaA los pacientes subsidiarios de este tratamiento debe dárseles una información detallada sobre las distintas modalidades, informándoles de los riesgos y beneficios, de los efectos secundarios, así como de la necesidad de tratamiento médico mientras la radioterapia es eficaz, ya que habitualmente el control de la enfermedad suele retrasarse varios años.
Existen 2 modalidades de radioterapia: la radioterapia convencional conformada (RTC) y la radioterapia estereotáxica (RE), bien en dosis única (radiocirugía, RC), bien en dosis múltiples (RE fraccionada, REF)78.
La RTC puede disminuir las concentraciones de GH y normalizar las de IGF-I en el 60% de los pacientes, pero la máxima respuesta puede conseguirse a los 10-15 años. Consigue el control del volumen tumoral en 85-90% de los pacientes, con descenso del volumen tumoral en más del 50% de los pacientes (GradoC). La progresión del tumor después de la radioterapia es excepcional. El tratamiento médico debe mantenerse mientras la radioterapia es eficaz (GradoB). Se administra habitualmente en dosis de 160-180cGy durante 4 o 5 días/sem durante 5 o 6 sem (dosis total 4.500-5.000cGy).
En la actualidad las técnicas RE son de elección, ya que permiten una mejor planificación del campo a radiar, con un menor riesgo de radiación de estructuras adyacentes79,80.
En pacientes con pequeños restos tumorales, alejados más de 5mm de la vía óptica y con concentraciones no excesivamente elevadas de GH e IGF-I, debe recomendarse tratamiento con RC (gamma-knife, acelerador lineal, protones, etc.) (GradoC). La RC consigue habitualmente un control bioquímico más precoz. Se han reportado rangos de remisión del 17-50% en un periodo de seguimiento de 2 a 5 años. Algunas series repiten RC cuando no se consigue remisión tras la primera dosis. El tiempo de repetir el tratamiento o el incremento de las complicaciones no está establecido8,9,79–81.
En pacientes con restos tumorales grandes, con invasión de estructuras vecinas y concentraciones elevadas de GH e IGF-I, sobre todo si no responden adecuadamente a tratamiento médico, debe aconsejarse REF8,9,78,79.
Tratamiento médico durante la radioterapiaAlgunos estudios recomiendan suspender el tratamiento médico con AASS o AD al menos un mes antes y durante el tiempo que dure la radioterapia, debido a su efecto radioprotector82,83, pero otros estudios no confirman estos hallazgos84. No existen datos concluyentes en este sentido, ni está bien definido cuándo debería suspenderse el tratamiento antes de la radioterapia (GradoC).
SeguimientoTras la radioterapia el paciente debe continuar con el tratamiento médico para conseguir un adecuado control, mientras esta es eficaz. Se aconseja disminuir la dosis y suspender de forma periódica el tratamiento médico con objeto de evaluar el control de la enfermedad (GradoB).
Anualmente debe realizarse estudio de función hipofisaria (gonadal, tiroideo y adrenal) para iniciar tratamiento sustitutivo si el paciente presenta hipopituitarismo (GradoB)9.
ComplicacionesEl hipopituitarismo a largo plazo aparece en más del 50% de los pacientes a los 5 años (GradoA). La incidencia y severidad es dependiente de la dosis. La incidencia de hipopituitarismo en la mayoría de las series tras RC es similar a la de RTC. La presencia de hipopituitarismo se asocia a un incremento de riesgo de mortalidad. En pacientes jóvenes que deseen fertilidad, debe considerarse el riesgo de hipopituitarismo antes de recomendar la radioterapia (GradoB).
La radioterapia se asocia también a incremento del riesgo de mortalidad y de enfermedad cerebrovascular. El riesgo es dependiente de la dosis. En un estudio reciente de Sherlock et al. de 501 pacientes acromegálicos tratados con radioterapia (n=237) se observa un incremento de todas las causas de mortalidad frente a los pacientes no radiados (n=264). El riesgo de muerte por enfermedad cerebrovascular fue 4 veces superior en los pacientes irradiados. Pérdida de visión se presenta en algunos casos (0-3%), es debida a neuritis óptica, en relación con la dosis total acumulada85
La radioterapia también se ha asociado a la aparición de segundos tumores y radionecrosis y se presenta en el 2% de los pacientes tratados con RTC. No hay datos sobre incremento de riesgo de mortalidad o de enfermedad cardiovascular en pacientes tratados con RC. En una serie de pacientes tratados con RC la incidencia de radionecrosis fue del 0,8%, si bien algunos pacientes habían sido previamente tratados con RTC. El riesgo de deterioro cognitivo tras radioterapia continúa siendo controvertido (GradoC)9.
Comorbilidades de la acromegaliaLas comorbilidades asociadas con los tumores productores de GH pueden ser debidas al efecto local del tumor, a los déficits hormonales hipofisarios asociados o al exceso de GH e IGF-I. Respecto a las comorbilidades dependientes del exceso de GH o IGF-I, una detección precoz y una corrección de los factores de riesgo asociados nos permitirían reducir la mortalidad secundaria a estas complicaciones. Aunque el control bioquímico de la acromegalia puede mejorar o estabilizar estas comorbilidades, un porcentaje significativo de pacientes van a precisar tratamiento adicional8,9,86,87.
Complicaciones cardiovasculares y factores de riesgo cardiovascularHipertensión arterialLa hipertensión arterial es muy prevalente (superior al 40%) en pacientes con acromegalia, por ello el control tensional debe realizarse de forma rutinaria. La hipertensión arterial se agrava por la apnea del sueño. El tratamiento de la hipertensión arterial debe ser precoz y agresivo, independiente del tratamiento de la acromegalia y con un objetivo de alto riesgo: mantener valores tensionales inferiores a 130/80mmHg (GradoA, BEL3). El tratamiento específico de la hipertensión arterial en estos pacientes no está bien definido9,87.
MiocardiopatíaLa hipertrofia biventricular es el hallazgo característico de la cardiopatía acromegálica. Inicialmente se expresa como disfunción diastólica de esfuerzo, que puede progresar a disfunción diastólica en reposo y rara vez a miocardiopatía congestiva dilatada. Pueden aparecer arritmias en un 40% de los pacientes87.
Valoración: se debe valorar el riesgo cardiovascular incluyendo colesterol total, c-LDL, c-HDL y triglicéridos (GradoC)9,87. La valoración cardiológica debe incluir electrocardiograma. Se recomienda realizar ecocardiograma si hay evidencia de hipertrofia de ventrículo izquierdo en el electrocardiograma o si tienen síntomas que incluyen arritmias o disnea (GradoC)9,87.
Los pacientes con acromegalia, especialmente aquellos con gigantismo, requieren valoración del sistema arterial periférico, incluyendo enfermedad venosa periférica.
Efectos posibles del tratamiento de la acromegalia: la disminución de las concentraciones de GH puede mejorar el tamaño del ventrículo izquierdo y la función cardíaca (BEL3). No obstante, la función cardíaca se normaliza solo en el 50% de los pacientes mayores de 40 años9,87.
Los posibles efectos valvulares de la cabergolina en dosis altas en pacientes con acromegalia son controvertidos, no obstante, se debe solicitar un ecocardiograma antes de iniciar este tratamiento y repetir en intervalos regulares posteriormente.
Tratamiento de las complicaciones cardíacas: se debe usar tratamiento estándar para manejar la hipertrofia del ventrículo izquierdo, la alteración de la función diastólica o sistólica, las arritmias, las alteraciones valvulares o la enfermedad isquémica (GradoC).
Diabetes mellitusLa diabetes es un factor predictor importante en el aumento de mortalidad en los pacientes con acromegalia, por ello, se debe monitorizar y realizar tratamiento adecuado (GradoC). En general, el descenso de GH mejora el control glucémico, independientemente del tratamiento utilizado.
La diabetes mellitus debe tratarse igual que en los pacientes diabéticos no acromegálicos, con el objetivo de mantener una hemoglobina A1c inferior a 6,5% (GradoC)9.
En algunos pacientes cuyo control glucémico se deteriora durante el tratamiento con AASS se puede valorar cambiar la medicación a PEG9.
Complicaciones respiratoriasApnea del sueñoEl síndrome de apnea del sueño (SAS) tiene una alta prevalencia en la acromegalia, afectando hasta al 70% de los pacientes de reciente diagnóstico. La causa principal es la obstrucción por engrosamiento faríngeo y macroglosia, aunque puede tener también un componente central. Se recomienda, si hay síntomas evidentes, realizar una oximetría domiciliaria y, según resultados, una polisomnografía nocturna (GradoC)9,87.
El tratamiento de la acromegalia reduce los tejidos blandos y puede hacer remitir el SAS (EL3), aunque es frecuente su persistencia. En este caso se deben utilizar otras medidas terapéuticas, incluyendo la respiración asistida (CPAP) (por ejemplo, medidas especiales para acromegalia como piezas bucales especializadas). Se recomienda también consulta con un cirujano maxilofacial (GradoC)9.
Los pacientes con obstrucción de las vías aéreas superiores por deformidad mandibular, macroglosia o hipertrofia de epiglotis pueden tener complicaciones durante la anestesia. Esto debe ser considerado de forma preoperatoria (GradoC)9.
Complicaciones osteoarticulares y dentalesLas alteraciones óseas y articulares no suelen ser reversibles tras la curación de la acromegalia (BEL2). Se recomienda por ello que cualquier procedimiento quirúrgico se posponga hasta la normalización de los niveles de GH e IGF-I (GradoD)9.
ArtropatíaLa degeneración articular puede afectar a cualquier articulación y los cambios son, en general, irreversibles (EL3). Se debe intentar realizar un diagnóstico y tratamiento precoz y debería manejarse de forma agresiva con fisioterapia, antiinflamatorios sistémicos o intraarticulares, o implantación de una prótesis cuando sea necesaria (GradoC)9.
Síndrome del túnel carpianoDebe valorarse regularmente la presencia de síntomas y/o signos de síndrome del túnel del carpo. Los síntomas pueden mejorar al reducir la GH (EL2), pero si persiste debe realizarse tratamiento específico, incluyendo el quirúrgico (GradoC)9.
Hipercalciuria e hipercalcemiaPueden aparecer por exceso de GH, se deben a una alteración del metabolismo de la vitamina D, y son reversibles con la curación de la acromegalia (EL3). Si persiste la hipercalcemia hay que descartar hiperparatiroidismo concomitante en el contexto de neoplasia endocrina múltiple9.
OsteoporosisLos pacientes con acromegalia deben ser evaluados en cuanto a factores de riesgo de osteoporosis. Se recomienda realizar densitometría ósea y determinación de los niveles de calcio en el momento del diagnóstico, sobre todo si hay historia de hipogonadismo o fractura. La utilidad de la DEXA como técnica diagnóstica de osteoporosis no está bien documentada en pacientes acromegálicos, ya que el mayor grosor del hueso puede influir en la medición de la densidad mineral ósea.
Si hubiese osteoporosis y no mejorase con la corrección del hipogonadismo o el exceso de GH e IGF-I, se debe considerar tratamiento antirreabsortivo (GradoC)9.
Pólipos y cáncer de colonAunque no hay suficiente evidencia para decir que los pacientes con acromegalia tengan riesgo elevado de cáncer de colon, sí hay una mayor prevalencia de pólipos de colon. También se ha visto una mayor mortalidad por cáncer de colon de la esperada (ratio 2,47). Por ello, se recomienda realizar una colonoscopia de cribado en el momento del diagnóstico en adultos (GradoC, BEL3). Si no hubiese pólipos ni cáncer, se recomienda revisar igual que a la población general con riesgo elevado (colonoscopia cada 5 años). Si la colonoscopia fuese anormal, hay que seguir las guías clínicas de seguimiento específicas9,87.
Complicaciones psicosocialesLa acromegalia se asocia con un deterioro de la calidad de vida (EL3), y los tests específicos de medición de esta variable, como AcroQoL, son una importante herramienta para la medida de resultados y se debe utilizar en la práctica clínica.
El control bioquímico mejora varios aspectos de la función psicosocial (EL3), aunque la normalización bioquímica no siempre normaliza estos aspectos9.
MortalidadLa acromegalia se asocia con un incremento de la mortalidad de 2 a 2,5 veces. La normalización de GH o IGF-I puede mejorar el riesgo de mortalidad (BEL2)88. Los niveles de GH (medidos por métodos sensibles) inferiores a 1 tienen una mortalidad similar a la esperada (EL2).
Seguimiento de la acromegalia a largo plazoDebe realizarse en función del grado de control de la enfermedad (tabla 3):
- 1.
Acromegalia curada tras cirugía: tras confirmación de control en postoperatorio (3-6 meses poscirugía), con normalización de GH e IGF-I y supresión de GH tras SOG (inferior a 1 o 0,4μg/l), los pacientes deben ser seguidos de forma periódica. Deberán realizarse determinaciones seriadas de las concentraciones de GH e IGF-I (cada 6 meses al inicio y anualmente después). Si hubiese criterios de recidiva, deberá realizarse SOG para confirmación.
- 2.
Acromegalia controlada con tratamiento médico: en pacientes tratados con AASS deberán monitorizarse con determinación de GH e IGF-I, inicialmente cada 3 o 4 meses para ajuste de dosis y, posteriormente, bianual o anualmente. No es necesaria la realización de SOG en el seguimiento. Tras control adecuado, se puede alargar el intervalo de dosis de análogos o disminuir esta. En pacientes tratados con PEG la monitorización deberá realizarse solo con la determinación de IGF-I.
- 3.
Acromegalia tratada con radioterapia: la monitorización debe realizarse igual que en los pacientes en tratamiento médico. Tras control adecuado deberá disminuirse la dosis del fármaco o suspenderlo a fin de confirmar la curación definitiva. También deberá monitorizarse la función hipofisaria de forma periódica por si aparece hipopituitarismo.
La RM deberá realizarse a los 3 o 4 meses de la cirugía y a los 3 o 4 meses del inicio del tratamiento médico. Los controles posteriores dependerán del control de la enfermedad; si el paciente está curado tras la cirugía no sería necesario realizar más RM, aunque esto no está claramente establecido; una alternativa podría ser realizar una RM cada 2-3 años, o antes si hay recurrencia clínica o bioquímica. En pacientes controlados con AASS deberá realizarse una RM al año con objeto de comprobar la disminución del tamaño tumoral. La frecuencia posterior no está establecida y dependerá del criterio clínico. Si el paciente no está controlado deberá realizarse cada 6 meses al inicio y anualmente después. En pacientes en tratamiento con PEG deberá realizarse cada 6 meses al inicio por si hubiese crecimiento tumoral y anualmente después. En pacientes tratados con radioterapia deberá realizarse control anual hasta la desaparición o el control tumoral.
Otro aspecto importante en el seguimiento a largo plazo es el control de las comorbilidades y, en los pacientes irradiados, el control de la función hipofisaria para iniciar tratamiento sustitutivo si aparece hipopituitarismo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.
