





La enfermedad celíaca es un trastorno inflamatorio del intestino delgado inducido por la ingestión de gluten de trigo y otras prolaminas en individuos genéticamente susceptibles, que se manifiesta por linfocitosis intraepitelial y de lámina propia, pérdida de vellosidades, remodelación tisular y presencia de anticuerpos antitransglutaminasa. El modelo patogénico más aceptado depende de la inmunidad adaptativa tras la estimulación de linfocitos T CD4+ por péptidos de gluten modificados por la transglutaminasa tisular y restricción por moléculas HLA-DQ2/DQ8, que producen citocinas proinflamatorias. El gluten activa también la inmunidad innata y la citotoxicidad epitelial mediada por linfocitos intraepiteliales. Aunque no está claro aún cuál es el efecto de los anticuerpos específicos, la disponibilidad de marcadores serológicos e inmunogenéticos como herramientas diagnósticas ha propiciado el avance en el conocimiento de la enfermedad celíaca y la revisión de los criterios diagnósticos, especialmente en los individuos adultos con expresión mínima o atípica de la enfermedad.
Celiac disease is an inflammatory disorder of the small intestine induced by intake of wheat gluten and other prolamines in genetically susceptible individuals. This disease is manifested by an increased number of intraepithelial and lamina propria lymphocytes, villous atrophy, tissue remodeling and the presence of anti-transglutaminase antibodies. The most widely accepted pathogenic model is based on adaptive immunity after T CD4+lymphocyte stimulation by tissue transglutamine-modified gluten peptides and HLA-DQ2/DQ8 restriction, which produce proinflammatory cytokines.
Gluten also activates innate immunity and epithelial cytotoxicity mediated by intraepithelial lymphocytes. Although the effect of specific antibodies remains unclear, the availability of serological and immunogenetic markers as diagnostic tools has increased our knowledge of celiac disease and has led to a reevaluation of the diagnostic criteria, especially in adults with minimal or atypical disease expression.
La enfermedad celíaca (EC) es un modelo muy interesante para estudiar cómo la interacción entre factores genéticos y ambientales lleva a la pérdida de tolerancia oral del sistema inmunitario a una proteína de la dieta, como es el gluten1,2. Se ha avanzado mucho en el conocimiento de la enfermedad molecular de esta enfermedad, en especial con la identificación de los heterodímeros HLA-DQ2 y DQ8, su papel en la presentación de gluten a los linfocitos T CD4+ específicos3 y de la acción directa de ciertos fragmentos de las gliadinas sobre el epitelio4–6. La inflamación de la mucosa y el desarrollo de la lesión intestinal son secundarios a la activación secuencial y la superposición de las respuestas inmunitarias innata y adaptativa, que conducen a la alteración de la producción local de citocinas por los linfocitos T locales1,2.
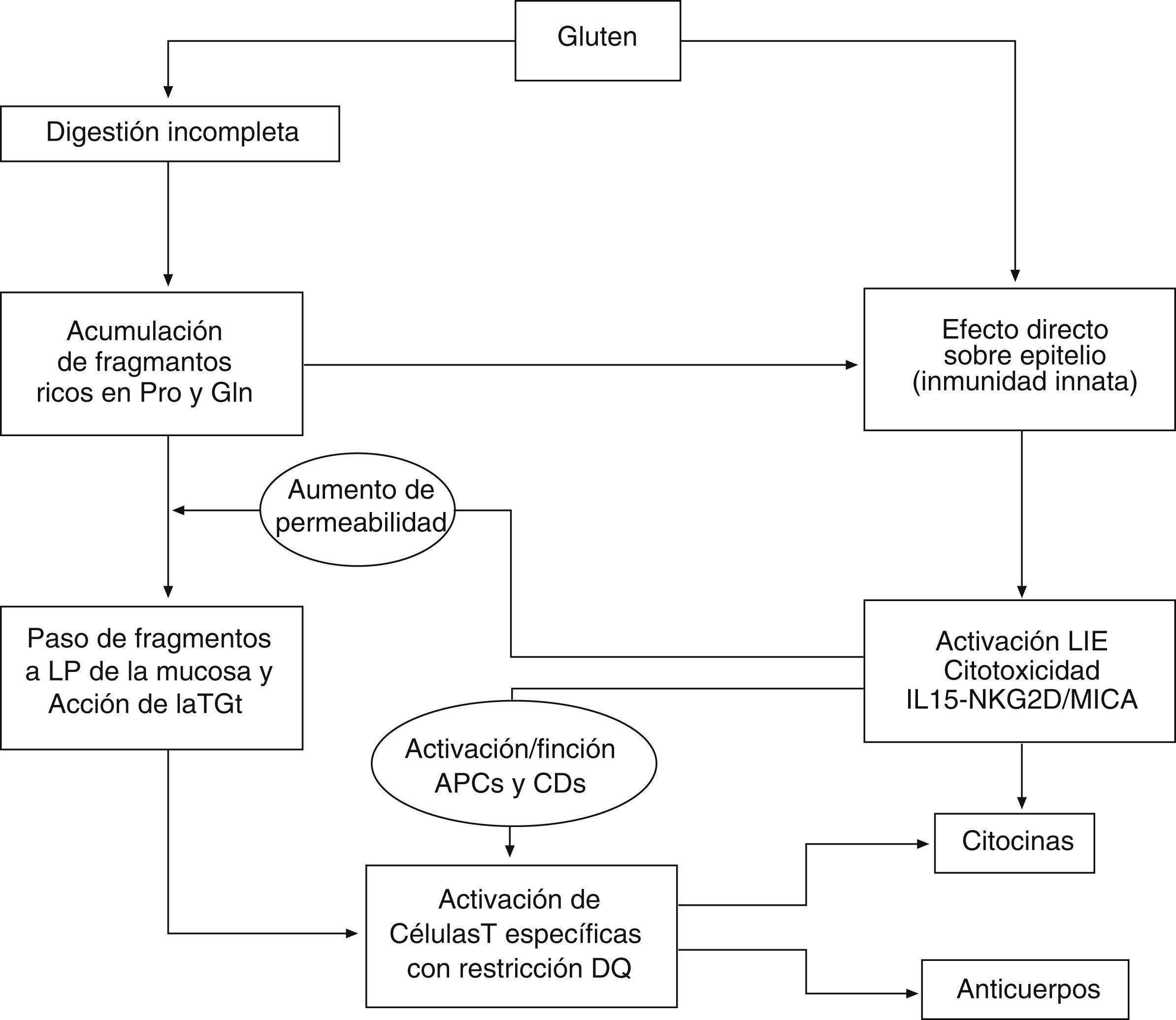
El modelo patogénico más aceptado integra factores que actúan tanto en el epitelio como en la lámina propia, como la digestión incompleta y el transporte transepitelial de péptidos7,8, el efecto tóxico directo del gluten sobre el epitelio, la proliferación y la activación de linfocitos intraepiteliales (LIE)1,6, y el reconocimiento de péptidos de gluten por linfocitos T específicos con restricción HLA-DQ2 tras ser modificados por la transglutaminasa tisular (TGt o TG2)3,9 (fig. 1). La principal laguna en el conocimiento de la patogenia de la EC es explicar por qué solo unos pocos individuos portadores del human lymphocyte antigen (HLA) de riesgo desarrollan la enfermedad. Es posible que otros factores no solo genéticos, sino también ambientales (por ejemplo, la composición de la flora intestinal) influyan en la capacidad individual de inducción y control de la respuesta innata, y en la susceptibilidad del individuo.

Las principales familias de proteínas del gluten de trigo (gliadinas y gluteninas), y sus homólogos en la cebada y centeno, denominadas prolaminas por su alto contenido en los aminoácidos glutamina y prolina10, contienen fragmentos nocivos para el intestino celíaco. Se han identificado 2 tipos de péptidos: inmunogénicos, que estimulan linfocitos T del intestino o sangre periférica de los pacientes celíacos con restricción DQ2/DQ8, y pueden ser epítopos inmunodominantes (como los residuos 57–75 de alfa-gliadina)9,11–13; y péptidos tóxicos (residuos 31–43/49) de acción directa sobre el epitelio, que son independientes de los linfocitos T1,5. No todos los cereales contienen la misma proporción de péptidos de cada tipo, ni la misma cantidad relativa de gluten, de ahí las variaciones en su capacidad patogénica: la avena tiene alrededor del 10% de contenido de gluten que tiene el trigo.
La EC está fuertemente asociada con genes HLA (locus CELIAC1, cromosoma 6p21): la mayoría de los pacientes celíacos muestra una variante de la molécula HLA-DQ2 codificada por los alelos DQA1*05 y DQB1*02, y el resto son DQ8 (DQA1*03, DQB1*0302), o son portadores de algún alelo aislado del DQ214,15. Estos genes muestran un efecto dosis mediado por una presentación de péptidos, que es más eficaz en los homocigotos HLA-DQ2. Aunque el 25% de la población es portadora de DQ2, solo el 1% desarrolla la EC1,14. Estudios de genoma completo han identificado otras regiones que incluyen genes de susceptibilidad, muchos de éstos relacionados con la función inmunitaria16, y que podrían compartirse también con otras enfermedades crónicas de base inmunológica, como la diabetes mellitus17.
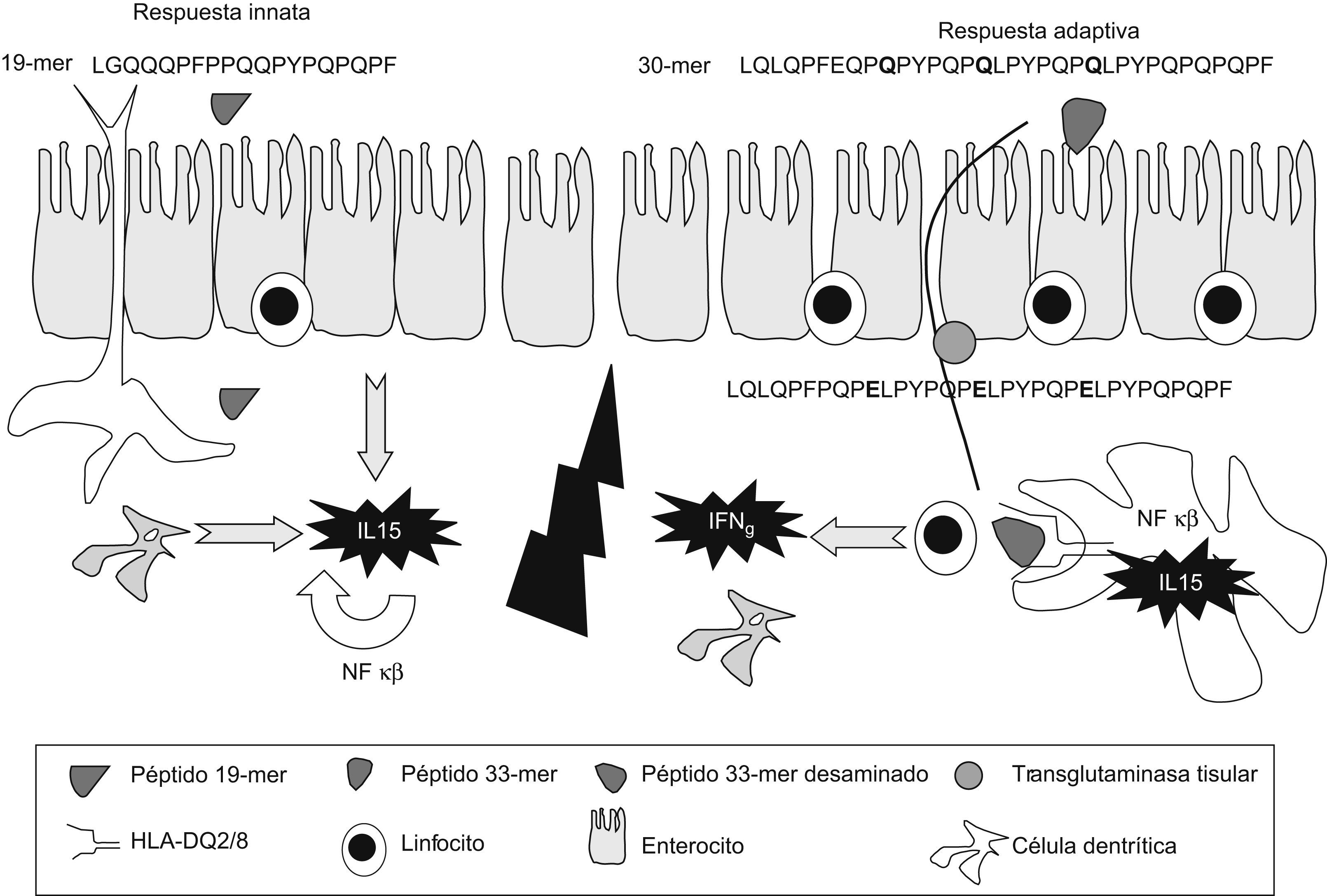
Paso de péptidos de gluten a través del epitelioLa principal forma de entrada de los péptidos de gliadina a través del epitelio es la transcitosis, aunque el mecanismo causante no está aclarado del todo2. En la EC activa se ha observado un aumento del transporte transepitelial dependiente del IFN-γ, pero el procesamiento de estos péptidos por las células del epitelio está también alterado, de forma que los péptidos tóxicos (19-mer) y los inmunogénicos (33-mer), tanto en forma intacta como parcialmente degradada, podrían pasar al interior7,8. Por otro lado, en situaciones de inflamación, como en la EC activa, se ha observado un aumento de la difusión paracelular pasiva de péptidos debido a la liberación de zonulina inducida por los péptidos de gliadina, que actúa sobre las uniones estrechas del epitelio18.
Otros mecanismos activos que podrían contribuir también al paso de péptidos intactos de las gliadinas a través del epitelio intestinal son los que dependen de la presencia de un receptor, por ejemplo, mediante una vía que utiliza al receptor de la transferrina CD71 expresado por los enterocitos, y que permite el paso de complejos formados por péptidos de gliadina y anticuerpos específicos de IgA secretora, protegidos de la degradación por las enzimas lisosomales19, o mediante la captación directa de estos péptidos por células dendríticas mieloides de la lámina propia mucosa que expresan en membrana moléculas HLA-DQ y TGt20.
Inmunidad innata frente al glutenLos péptidos de gluten, como el p31–43/49 de la alfa-gliadina, pueden dañar directamente el epitelio intestinal al activar mecanismos de la inmunidad innata, con producción de IL15 e inducción de apoptosis en los enterocitos, que alteran la función barrera epitelial y el aumento de la permeabilidad4,5,7. La IL15 induce la proliferación y la activación de LIE T CD8+ que expresan receptores de células asesinas naturales (NK) tipo NKG2D y CD94-NKG2A, cuyos ligandos son las moléculas de estrés MICA/B y HLA-E, respectivamente, expresadas por los enterocitos en situaciones de estrés21–23. La IL15 promueve en los LIE la producción del IFN-γ y la citotoxicidad dependiente de proteínas citolíticas, como perforinas o granzima24,25. La lesión del epitelio depende de los LIE que expresan el receptor de célula T-αβ, que descienden tras la restricción de gluten, mientras que los LIE receptores de célula T γδ NKG2A tendrían una función reguladora26, y se mantienen elevados en dieta sin gluten (fig. 2).
. Se activan respuestas de citocinas proinflamatorias y mecanismos causantes de la transformación mucosa.")
El gluten tiene un efecto doble sobre el intestino: algunos péptidos, como el fragmento p31–43 de la alfa-gliadina induce estrés de los enterocitos, que expresan IL15 y moléculas MICA, y activa a los linfocitos intraepiteliales para expresar el receptor NKG2D y activar la citotoxicidad contra la célula epitelial. El paso de péptidos inmunogénicos a la lámina propia estimula a linfocitos T CD4+ específicos cuando se presentan junto a moléculas HLA-DQ2/DQ8, tras tener una modificación por parte de la transglutaminasa tisular (TGt o TG2). Se activan respuestas de citocinas proinflamatorias y mecanismos causantes de la transformación mucosa.
Recientemente, se ha conocido que los péptidos de gliadina captados por las células epiteliales mediante endocitosis son capaces de llegar hasta las vesículas paranucleares de estas células (endosomas tardíos y lisosomas); sin embargo, en vez de ser degradados en los lisosomas, el péptido p31–43 se acumula allí por causas aún desconocidas, lo que provoca un microambiente prooxidativo que induce la activación de la TGt y la degradación de la peroxisome proliferator-activated receptor (PPARγ), que es una molécula capaz de modular la inflamación intestinal. Este mecanismo podría explicar por qué los pacientes celíacos recaen tras la reintroducción del gluten, incluso antes de que aparezcan signos de inflamación27.
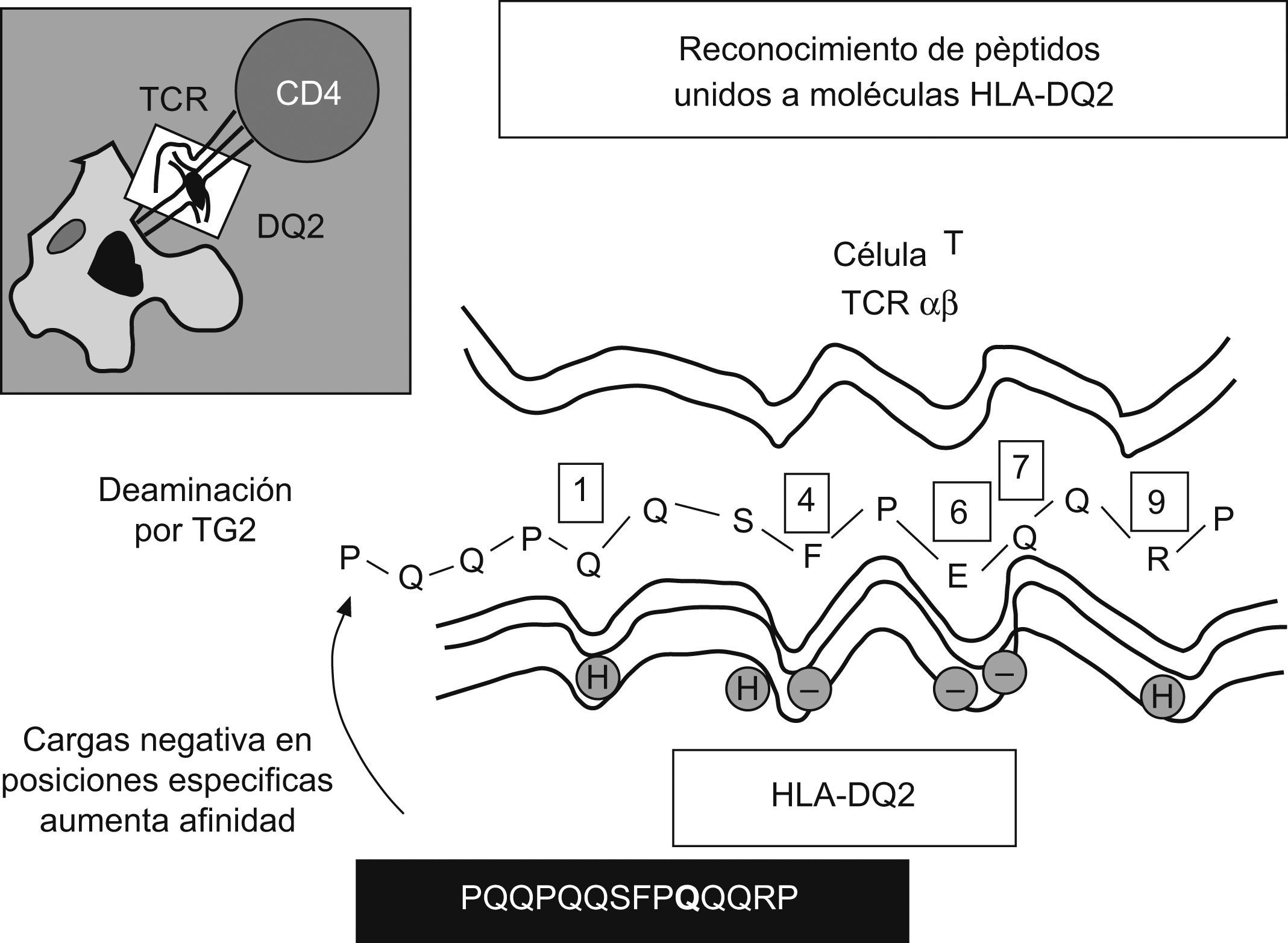
Respuesta inmunitaria adaptativa frente al glutenLos linfocitos T CD4+ específicos de la lámina propia reconocen péptidos sólo cuando se presentan mediante células dendríticas junto a moléculas HLA-DQ2/DQ83,28. Estas moléculas disponen de un bolsillo de unión a péptidos con propiedades únicas para acomodar secuencias peptídicas: DQ2 tiene preferencia por aminoácidos de carga negativa en posiciones centrales (P4, P6 o P7), y en el caso de DQ8, las posiciones son más externas (P1 o P9)29. De forma natural, las proteínas del gluten tienen pocas cargas negativas; sin embargo, la TGt liberada durante la inflamación es capaz de inducir la conversión de residuos de glutamina en ácido glutámico en secuencias del tipo glutamina, prolina u otro aminoácido30,31 (fig. 3).
 de la alfa-gliadina y la molécula HLA-DQ2, con posiciones de anclaje4,6,7 que tienen preferencia por cargas negativas. La transglutaminasa tisular induce la sustitución de glutamina de carga positiva por ácido glutámico de carga negativa (glutamina, ácido glutámico o prolina).")
La molécula HLA-DQ sirve de elemento de restricción en el reconocimiento de epítopos de gluten por los linfocitos T CD4+. Representación esquemática de la interacción entre un epítopo (PQQPQQSFPQQRP) de la alfa-gliadina y la molécula HLA-DQ2, con posiciones de anclaje4,6,7 que tienen preferencia por cargas negativas. La transglutaminasa tisular induce la sustitución de glutamina de carga positiva por ácido glutámico de carga negativa (glutamina, ácido glutámico o prolina).
Precisamente por su alto contenido en glutamina y prolina, los péptidos de gluten son resistentes a la proteolisis por enzimas digestivas, y se forman fragmentos grandes que contienen varios motivos del tipo glutamina, prolina u otro aminoácido, que son los substratos preferidos de la TGt32,33, como el péptido de 33 aminoácidos (p57–89 de la alfa-gliadina) que contiene 6 copias de 3 epítopos T, y cuya inmunogenicidad para los linfocitos T del intestino celíaco aumenta tras la deamidación por TGt8. La activación de estos linfocitos T CD4+ reactivos al gluten conduciría una respuesta proinflamatoria dominada por la producción de IFN-γ. Algunas enzimas bacterianas, como la prolil-endopeptidasa, inducen la degradación de estos fragmentos e impiden que se formen epítopos T capaces de activar respuestas de la inmunidad adaptativa34.
La respuesta adaptativa humoral parece tener una contribución directa menor en la patogenia de la EC, aunque (ver más arriba) los anticuerpos de IgA secretora específicos de gliadina podrían favorecer la translocación o el paso de péptidos de gliadina intactos desde la luz intestinal hasta el interior por una vía en la que intervendría el receptor de la transferrina CD712. No se han identificado linfocitos T específicos de TGt, y falta por explicar aún por qué la ingestión de una proteína de la dieta (gluten) promueve la producción de autoanticuerpos frente a la TGt1,3. Como posible explicación se ha propuesto un modelo en el que los linfocitos T CD4+ reactivos al gluten podrían proporcionar la ayuda necesaria a las células B específicas de TGt para la síntesis de anticuerpos vía formación de complejos formados por TGt y péptidos de gliadina35.
Alteración de la red de citocinas y mediadores de inflamaciónLa tolerancia a los antígenos de la dieta se relaciona con la activación de células T reguladoras y la producción de citocinas de efecto inmunosupresor (IL10 o TGFβ) que previenen las respuestas inadecuadas de los linfocitos Th efectores36. En la EC activa los linfocitos T CD4+ de la lámina propia y los LIE CD8+ contribuyen a desencadenar una respuesta Th1 dominada por el IFN-γ, el factor de transcripción T-bet y otras citocinas proinflamatorias (TNF-α, IL18, IL21), junto a un descenso de IL10 e TGFβ37–39, y la producción de IL15 por los enterocitos5. Este perfil de tipo proinflamatorio, que desaparece en los pacientes en remisión, activa mecanismos efectores del daño tisular, en los que interviene el factor de crecimiento de queratinocitos40, y metaloproteinasas de matriz41,42, implicados en la degradación de la matriz extracelular y la transformación mucosa.
Al contrario de otras enfermedades inflamatorias crónicas del intestino, en la EC activa no se describe un aumento de IL12, principal citocina inductora de la respuesta Th1, por lo que otras citocinas deberían inducir la diferenciación Th1, como el IFN-α, producido por células dendríticas plasmacitoides43,44, o la IL2145,46, cuyo gen se localizó en una región ligada a la susceptibilidad de la EC16. Estas citocinas producidas por células de la inmunidad adaptativa (IFN-γ, IL21), o innata (IFN-α, IL15), podrían determinar el desarrollo de la inflamación y la enteropatía en la EC246, además de contribuir a la pérdida de tolerancia al gluten, debido al bloqueo de la vía de señalización del TGFβ, por la IL1547, o por inhibición de la supresión de los linfocitos T efectores por linfocitos T reguladores, a través de la IL212.
La inmunología en el diagnóstico y el seguimiento de la enfermedad celíacaDe acuerdo a los criterios de la ESPGAN, el diagnóstico de la EC se basa en los hallazgos anatomopatológicos de la biopsia intestinal cuando el paciente toma una dieta que contiene gluten, y la normalización clínica e histológica tras una dieta de exclusión. En relación con la ingestión de gluten, se ha observado todo un espectro de cambios histopatológicos e inmunológicos en el intestino. Los individuos con enteropatía mínima y síntomas asociados al gluten, o con alteraciones inmunológicas similares a las de los pacientes celíacos, pero sin histología o clínica definida, suponen un reto para el diagnóstico de la enfermedad. Por esta razón, en los últimos años, hubo un gran interés en la identificación de pruebas serológicas que permitieran la detección de individuos con expresión mínima o atípica de la enfermedad.
Marcadores serológicosLos anticuerpos antigliadina (AAG) no son específicos de la EC, se detectan también en otras enfermedades, e incluso en controles normales. No está clara su relación con la patogenia de la EC y pueden reflejar el aumento de la permeabilidad intestinal, como indica la producción de anticuerpos contra otras proteínas de la dieta, y su aparición en otras enteropatías48. Los AAG séricos se elevan en la EC activa, en paralelo con la ingestión de gluten, pero mientras los niveles de AAG-IgA disminuyen rápidamente tras retirar el gluten hasta hacerse negativos, los de clase IgG tienen una cinética más lenta y, en algunos casos, no llegan a desaparecer49. La edad influye en los niveles de AAG, son más elevados en los pacientes <2 años, en especial los de clase IgA, y su valor diagnóstico disminuye en niños mayores y en los adultos50.
Tanto para el uso aislado de AAG-IgA, como de forma conjunta con AAG-IgG, los valores de sensibilidad y especificidad publicados se encuentran entre el 70 y el 80%, aunque hay una gran variabilidad entre los estudios51. En la actualidad, con los marcadores serológicos alternativos que disponemos, no parece estar justificado el uso de los AAG en el diagnóstico de la EC52. Las únicas situaciones en las que podría mantenerse la utilización de estos marcadores son el control de la dieta sin gluten y la falta de sensibilidad de los anticuerpos antiendomisio (AEm)-IgA por debajo de los 2 años de edad, aunque en estos casos, los AAG se ven superados por sus sucesores naturales, los anticuerpos antipéptidos deaminados de gliadina53.
La descripción del patrón de AEm-IgA en la dermatitis herpetiforme y la EC por inmunofluorescencia indirecta sobre cortes de esófago de mono54 o de cordón umbilical humano55 revolucionó el diagnóstico serológico de la EC, y se alcanzaron unas cifras de sensibilidad y especificidad superiores al 95%51. Además de la utilización de los tejidos de mono, el principal problema de estos marcadores es técnico, puesto que la inmunofluorescencia indirecta requiere la interpretación del observador, es, por tanto, subjetiva, y necesita de un entrenamiento especial y experiencia. Se pueden detectar anticuerpos de clase IgG e IgA, aunque es este último isotipo es el que tiene valor diagnóstico en la EC en pacientes competentes para IgA.
Los problemas técnicos de los AEm se resolvieron con el aislamiento e identificación del principal antígeno del endomisio (TGt)56 y las pruebas basadas en los anticuerpos anti-TGt (aTGt). El uso de antígenos humanos (de eritrocitos o recombinantes) mejoró considerablemente la especificidad, aunque siguen apareciendo algunos falsos casos positivos en hepatopatías (cirrosis biliar primaria, otras cirrosis hepáticas y hepatitis crónicas por virus C), en artritis reumatoide, psoriasis o enfermedad de Crohn57, por lo que en estas enfermedades es aconsejable validar los casos positivos para aTGt con el estudio de AEm. A pesar de todo, los anticuerpos aTGt-IgA presentan unos valores de sensibilidad cercanos al 100% y de especificidad entre el 89 y el 96%51.
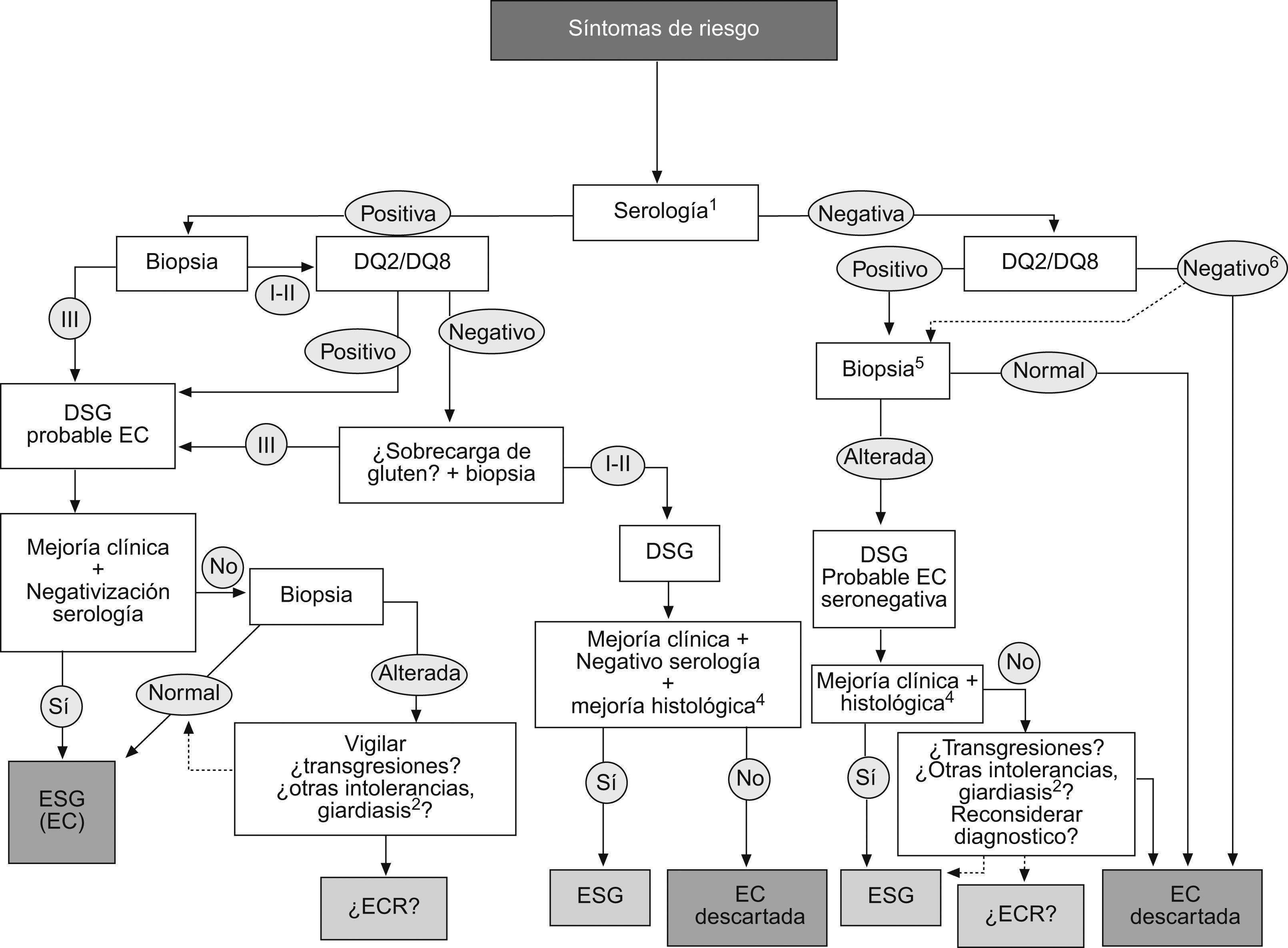
En general, los AEm-IgA parecen ser más específicos y los aTGt más sensibles, especialmente los que utilizan el antígeno recombinante humano58. Sin embargo, se ha observado una pérdida de sensibilidad en los casos de lesiones menores en la biopsia y pueden encontrarse casos de AEm-IgA negativos en un 69% de los pacientes adultos con alteraciones parciales (Marsh IIIa)59. Un patrón similar, aunque con mejores valores, se ha observado también para los aTGt-IgA60. Por el contrario, pueden encontrarse marcadores positivos sin alteración de la mucosa intestinal o con alteraciones leves (Marsh 0 a I)61. Para el control de la dieta, los aTGt-IgA pueden ser más adecuados que los AEm-IgA62, aunque se han perfilado otras alternativas, como los anticuerpos combinados IgA+IgG-aTGt62,63 o los anticuerpos antipéptidos deaminados de gliadina53. Sin embargo, es difícil determinar cuál es el mejor marcador en el seguimiento de la dieta, debido al menor número de biopsias de control que se realizan en este período (figs. 4).
. 2I, II y III: grados de la clasificación de Marsh, alteración de la mucosa intestinal. EC: enfermedad celíaca; ECR: enfermedad celíaca refractaria; ESG: enteropatía sensible al gluten; DSG: dieta sin gluten.")
Protocolo de uso de los marcadores de laboratorio en el diagnóstico de la enfermedad celíaca. 1Serología: anticuerpos anti-endomisio o antitransglutaminasa IgA (o IgG en déficit de IgA). 2I, II y III: grados de la clasificación de Marsh, alteración de la mucosa intestinal. EC: enfermedad celíaca; ECR: enfermedad celíaca refractaria; ESG: enteropatía sensible al gluten; DSG: dieta sin gluten.
En los últimos años se han publicado trabajos realizados en una población adulta, en los que se refleja que el valor de las pruebas serológicas de EC en los adultos es mucho menor de lo observado anteriormente en la población pediátrica, además de mostrar también la existencia de pacientes celíacos seronegativos64. Los estudios in vitro de producción de anticuerpos aTGt o AEm en cultivos de explantes de biopsias del intestino pueden resultar de ayuda para aclarar estos casos seronegativos, o para evitar la realización de pruebas de provocación in vivo65.
Marcadores inmunogenéticosLa asociación de la EC con la región HLA es una de las más fuertes descritas en cualquier enfermedad66. En la mayoría de las poblaciones estudiadas, más del 90% de los pacientes expresan el heterodímero HLA-DQ2 codificado por los alelos DQA1*05 y DQB1*02, en posición cis, asociados a DR3 (más común en el centro y norte de Europa), o en trans, en heterocigotos DR5/DR7 (que es más frecuente en la cuenca mediterránea). Sin embargo, DQ2 está presente en alrededor del 30% de la población general67. Del resto de los pacientes, muchos presentan un segundo heterodímero de riesgo DQ8 (codificado por los alelos DQA1*03 y DQB1*0302, en cis y asociado al DR4)68. Los pacientes que no expresan ni DQ2 ni DQ8 completos, son portadores de algunos de los alelos que codifican para el DQ2 por separado (DQA1*05 o DQB1*02)69. Entre los individuos portadores del DQ2, los que presentan una doble dosis del alelo DQB1*02 podrían tener un mayor riesgo de desarrollar la EC, como es el caso de los homocigotos DR3/DR3 y los heterocigotos DR3/DR770,71 (tabla 1).
Utilidad clínica de los marcadores genéticos de riesgo de enfermedad celíaca
| Hallazgos histológicos o serológicos dudosos |
| “EC latente”: AEm/TGt+ con biopsia normal |
| DSG sin biopsia previa |
| Exclusión de EC: síntomas con biopsia y serología normales |
| Selección de individuos de “alto riesgo” entre familiares o enfermedades asociadas |
AEm: anticuerpos antiendomisio; DSG: dieta sin gluten; EC: enfermedad celíaca; TGt: transglutaminasa tisular.
La concordancia del 30% entre hermanos con HLA idéntico, induce a pensar que en la susceptibilidad intervienen otros genes de dentro y de fuera de la región HLA. Además, menos del 2% de los portadores del DQ2 o DQ8 desarrollan la enfermedad, por lo que es probable la acumulación de riesgos por intervención de otros muchos genes de acción menor, y que podrían ser distintos para cada población o incluso para cada individuo. La mayoría de los pacientes DQ2 positivos son portadores del haplotipo ancestral 8.1 (B8-DR3-DQ2)72, que incluye otros alelos capaces de conferir riesgo o de modificar el efecto del DQ273. Este haplotipo está asociado también con otras enfermedades autoinmunitarias. Entre los posibles genes con implicación funcional incluidos en la región HLA estarían los genes del TNF-α74,75 y de la linfotoxina α. Se han encontrado asociaciones con otros genes de la región HLA, como los que codifican las moléculas MICA/B76, 77, y con moléculas de la familia de proteínas de estrés HSP-7078. La falta de replicación de estos hallazgos podría deberse a diferencias poblacionales en la contribución a la susceptibilidad.
Asociación con otras regiones genéticas distintas del HLAOtras zonas del genoma contienen también genes candidatos de participar en la susceptibilidad a la EC: 2p33: CELIAC3 (OMIM 609755), 5q31–33: CELIAC2 (OMIM 609754), 15q11–13: CELIAC5 (OMIM 607202) y 19p13.1: CELIAC4 (OMIM 609753). Estas zonas se han definido mediante estudios de análisis de ligamiento sistemáticos pangenómicos. La implicación de otras regiones (3p24, 9p21, 11p15, 11q23–q25, 17q22 y Xp11) no se ha reproducido en otras poblaciones. Los estudios de asociación son el abordaje para comprobar la implicación de genes candidatos en zonas calientes que, en la mayoría, contienen genes relacionados con la respuesta inmunitaria. En cada paciente se postula que diferentes combinaciones de las variantes de genes de efecto menor podrían determinar el curso o la expresión de la EC.
El gen CTLA4 (OMIM 123890) codifica un receptor que regula la activación de los linfocitos T conjuntamente con CD28 (OMIM 186760), ambos enmarcados (junto al locus ICOS [OMIM 604558]) en el cluster CELIAC3 (OMIM 609755) del cromosoma 2q33. Se ha investigado el efecto de esta región en otras enfermedades de naturaleza inflamatoria crónica o autoinmunitaria. Sin embargo, en la EC los resultados son contradictorios, algunos encuentran una asociación de este polimorfismo al desarrollo de la EC79, mientras que otros no confirman estos hallazgos.
En 19p13.11 se ha encontrado asociación con polimorfismos del gen MYOIXB (OMIM 602129) en una población holandesa80, aunque no se ha confirmado en otras poblaciones europeas, incluida la española. El cluster de los KIR, que se encuentra en 19p13.4, se ha asociado con la EC, así como el gen CD209 (OMIM 604672) en 19q13.3, con pacientes celíacos DQ2 negativos, en ambos casos en una población española. Esta zona genómica es también una región de susceptibilidad para la enfermedad inflamatoria intestinal (IBD6 [OMIM 606674]).
Además de estas zonas calientes del genoma, se ha encontrado asociación con la EC en otros genes y poblaciones determinadas: MBL2 (OMIM 154545), IFNG (OMIM 147570) en población española e italiana, IL6 (OMIM 147620) y MIF (OMIM 153620) en una población española, IL10 (OMIM 124092) en pacientes celíacos italianos con déficit de IgA, y con presentación precoz de la enfermedad en una población italiana, L-SELECTIN (OMIM 153240) en una población hindú, y MMP3 (OMIM 185250) en celíacos varones italianos.
En un estudio reciente de búsqueda pangenómica mediante una técnica de array del SNP con rastreo de más de 300.000 polimorfismos en una una amplia muestra de pacientes celíacos y controles16, se encontró que la única zona del genoma (aparte del HLA) con asociación significativa se localizaba en 4q27, en una zona en la que se encuentran los genes de la IL2 y de la IL21. Además, se observó un aumento de la expresión del ARNm de la IL21 en un grupo de pacientes con EC activa comparado con los controles. Posteriormente, sumando al estudio inicial varias colecciones de ADN europeas (1.643 pacientes y 3.406 controles) pudieron definir varios genes de moléculas relacionadas con el sistema inmunitario como posibles factores de riesgo de implicación funcional: CCR3, IL12A, IL18RAP, RGS1, SH2B3 y TAGAP, algunas compartidas también por la susceptibilidad a la diabetes mellitus tipo 181. Sin embargo, en este momento, sólo la IL21 podría tener un papel en la producción de la lesión mucosa45.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses

