




Las plaquetas
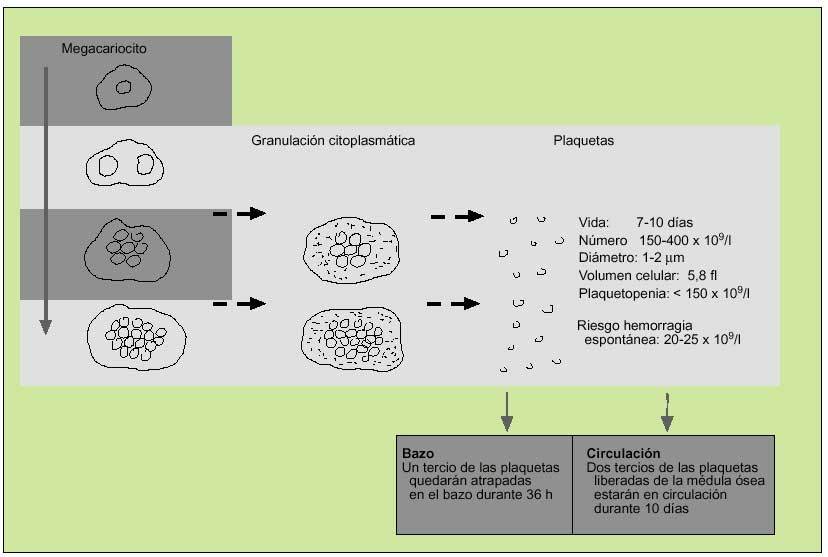
Como el resto de las líneas hematopoyéticas, las plaquetas se producen en la médula ósea, donde sus progenitores, los megacariocitos, madurarán a través de la replicación de su núcleo. A medida en que su número aumenta, el citoplasma se expande y cuando la cantidad de ADN se encuentra entre 16 y 32 veces más de lo normal, se granulará y las plaquetas serán liberadas. El precursor del megacariocito, el megacarioblasto, procede directamente de la diferenciación de la célula precursora hematopoyética, que habrá sido estimulada para diferenciarse por la acción de diferentes citocinas, entre las que se destaca la trombopoyetina (fig. 1).
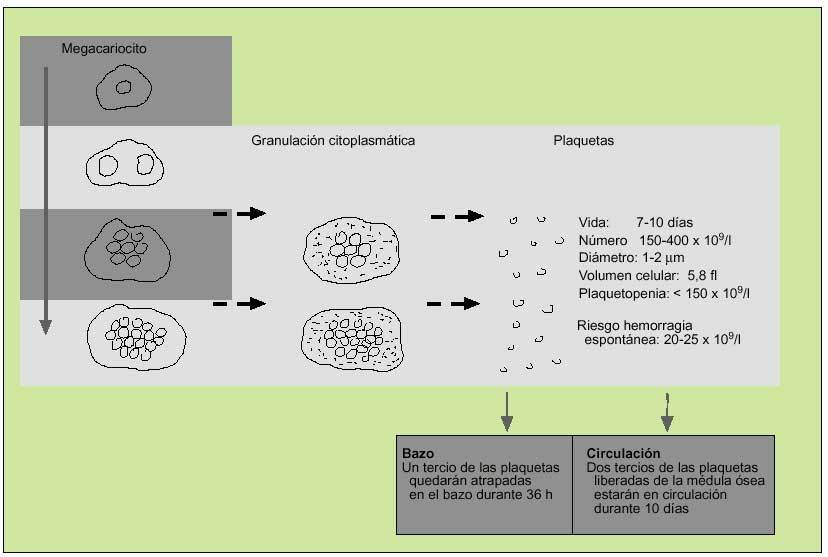
Fig. 1. Producción de plaquetas.
Tras abandonar la médula, todas las plaquetas se dirigen al bazo, donde una tercera parte son secuestradas y las restantes se liberan a la circulación. Posteriormente, tras 10 días son destruidas en el hígado por las células del sistema mononuclear fagocítico. Una célula madre dará lugar a la producción de 4.000 plaquetas en 10 días.
Morfología
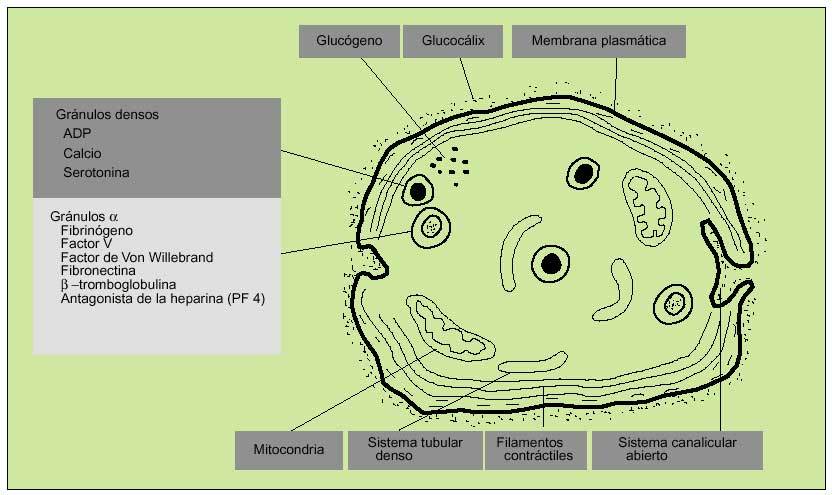
Las plaquetas se encuentran rodeadas por una cubierta glucoproteica que es fundamental en las reacciones de adhesión y agregación iniciales en el proceso de hemostasia primaria. En el interior de las plaquetas se diferencian dos tipos de gránulos: densos y alfa. Los primeros contienen calcio, ADP y serotonina; los segundos, factor plaquetario IV (antagonista heparínico), PDGF (protein derived growth factor), ß-tromboglobulina, fibrinógeno, FVW y otros factores de la coagulación. Otros organelos citoplasmáticos contienen enzimas hidrolíticas (lisosomas) y catalasas (peroxisomas).
El contenido de los gránulos se vierte en el sistema canalicular abierto (invaginación de la membrana plasmática).
El sistema reticular denso es probablemente el lugar de síntesis de prostaglandinas y tromboxano A2 (fig. 2).
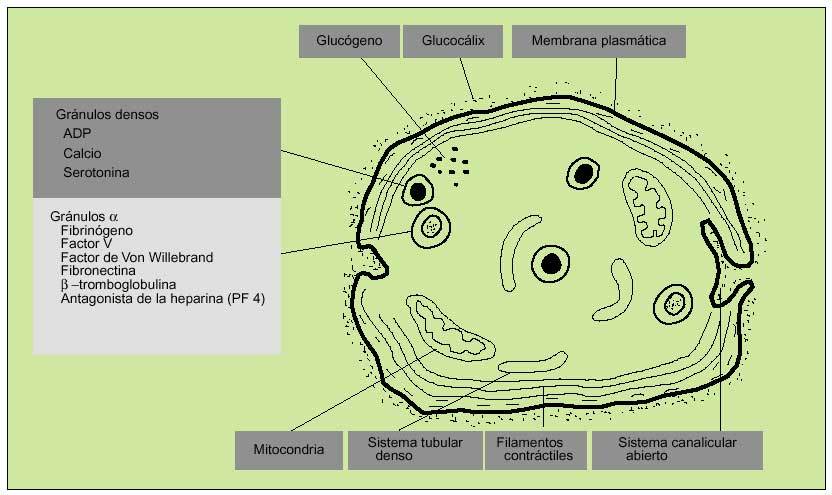
Fig. 2. Estructura de la plaqueta.
Función
Las plaquetas producen la formación de tapones mecánicos que detengan el sangrado en su fase aguda (hemostasia primaria). Tal acción requiere una secuencia de acontecimientos:
Adhesión
Una vez que se ha producido el daño vascular con la consiguiente extravasación de sangre, las plaquetas se adherirán al tejido conectivo del subendotelio expuesto.
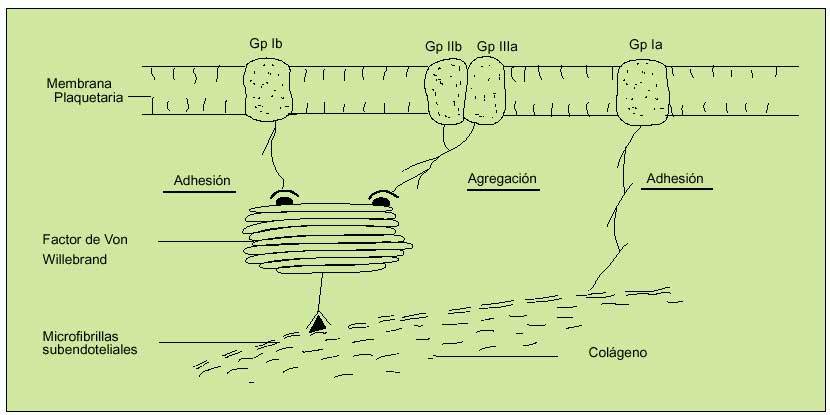
Los microfilamentos subendoteliales se unirán al FVW (liberado de las células endoteliales tras daño, estrés, ejercicio o estimulación adrenérgica), que formará así un puente con las plaquetas al unirse con la GpIb, estableciendo un primer nivel de anclaje. Esta unión dejará expuesto el receptor de la Gp IIb-IIIa en la superficie plaquetaria, que podrá unirse tanto al FVW de nuevo como al fibrinógeno. La adhesión directa al colágeno expuesto será mediada por una tercera glucoproteína, denominada Ia. De esta forma se logrará el anclaje de las plaquetas en el lugar del sangrado formando las primeras bases del tapón hemostático (fig. 3).
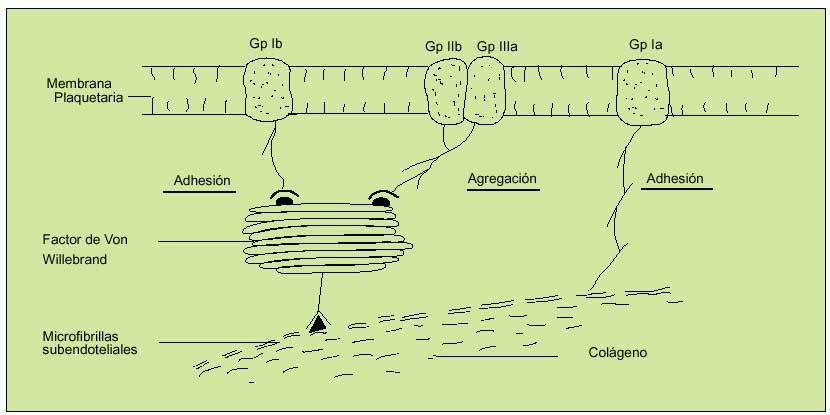
Fig. 3. Mecanismos de anclaje plaquetario. Hemostasia primaria. La unión inicial de la glucoproteína Ib al factor de Von Willebrand (FVW) define la fase inicial de adhesión. Esta unión activará al complejo glucoproteico Gp IIb/IIIa, que se unirá también al FVW y al fibrinógeno, constituyendo la fase de agregación. La GP Ia permitirá un anclaje plaquetario directo al colágeno.
La adhesión plaquetaria inducirá una serie de cambios metabólicos que darán lugar a un cambio estructural en las plaquetas, que adquirirán una forma esférica y emitirán seudópodos que estimularán la unión con plaquetas adyacentes.
Reacción de liberación
Tras la primera fase de adhesión, las plaquetas secretarán al exterior el contenido de sus gránulos: ADP, serotonina, fibrinógeno, enzimas, ß-tromboglobulina y factor plaquetario 4. Se liberará también ácido araquidónico desde la membrana celular que conducirá a la formación de tromboxano A2, que iniciará la reacción de liberación. El tromboxano A2 no sólo iniciará la agregación de las plaquetas, sino que inducirá una vasoconstricción local importante. La reacción de liberación estará contrarrestada por la prostaglandina PGI2, sintetizada por las células del endotelio vascular, potente inhibidor de la agregación y que probablemente ejerza un papel de prevención del depósito de plaquetas en el endotelio normal.
Fase de agregación
La liberación de ADP y tromboxano inducirá la agregación de las plaquetas de alrededor al foco de la lesión. El ADP inducirá la adhesión interplaquetaria. La propia agregación estimulará una mayor liberación de ADP y tromboxano, causando un feedback positivo que sólo se bloqueará una vez se haya obliterado la zona lesionada.
Los fosfolípidos de la membrana plaquetaria tienen un efecto procoagulante mediante la estimulación del factor X en Xa, así como en la formación de trombina a partir de la interacción del factor Xa, V y II (protrombina).
Trastornos de las plaquetas
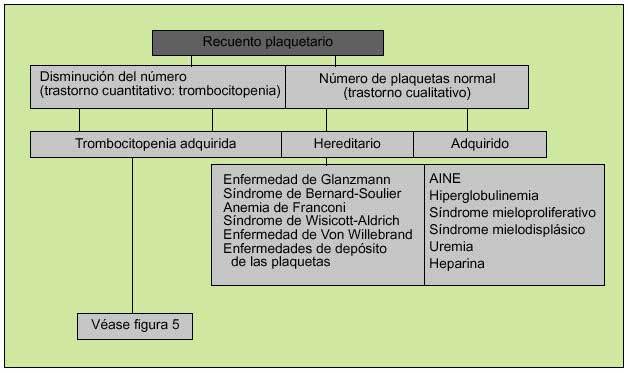
La función plaquetaria, como parte fundamental de la hemostasia primaria, podrá alterarse por disminución del número de éstas (plaquetopenia) o por déficits cualitativos que mermen sus propiedades (fig. 4). En el primer caso existirán dos circunstancias mayores que explicarán el descenso de su número, a saber: plaquetopenia central, en la que existirá un déficit de la producción de plaquetas y plaquetopenia periférica, ya sea por aumento de la destrucción de las plaquetas en circulación o por un excesivo secuestro esplénico. La plaquetopenia central puede ser selectiva (no afectar a otras líneas hematopoyéticas) o formar parte de un contexto de depresión medular más generalizado. Un número reducido de megacariocitos puede formar parte de una depresión medular generalizada en anemia aplásica, leucemia, mielodisplasia, posquimioterapia, posradioterapia.
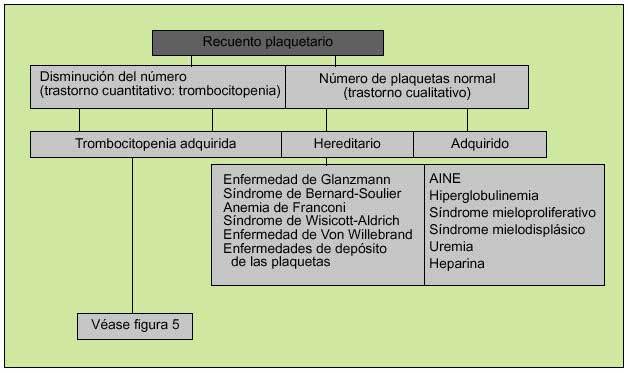
Fig. 4. Aproximación inicial a un problema de sangrado de origen plaquetario.
Sin embargo, en situaciones carenciales, como la anemia megaloblástica, el número de megacariocitos es normal o incluso elevado, fallando en este caso la producción de plaquetas desde el precursor medular.
Hay que tener presente que en la práctica clínica la causa más frecuente de trombopenia es la denominada seudotrombopenia, que se debe a un error de laboratorio debido a la agregación de las plaquetas in vitro inducida por el anticoagulante de la muestra, generalmente EDTA, por lo que debe confirmarse el diagnóstico mediante nueva extracción, esta vez en citrato.
La infección viral es la causa más común de trombopenia transitoria leve.
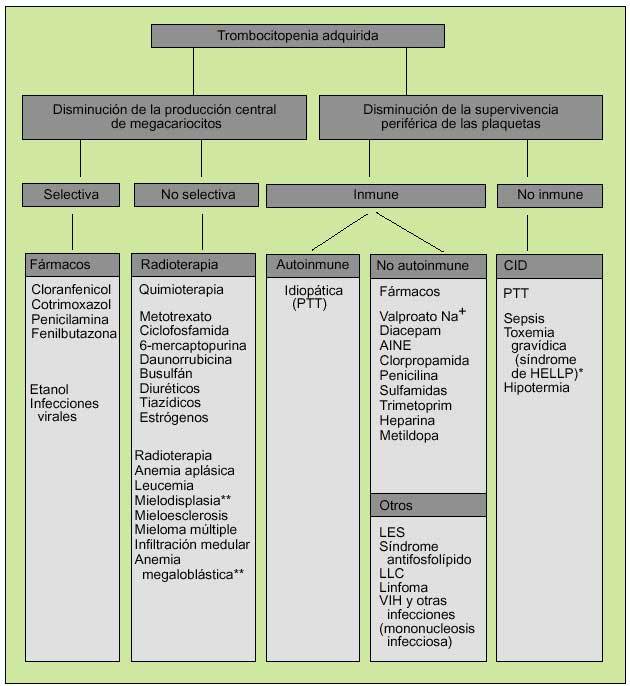
En cuanto a los trastornos cualitativos, éstos podrán ser adquiridos (la causa más común es la debida al uso de AINE) o hereditarios, debidos a la presencia de anticuerpos contra glucoproteínas específicas de superficie o a la disminución del FVW (tabla 1) (fig. 5).

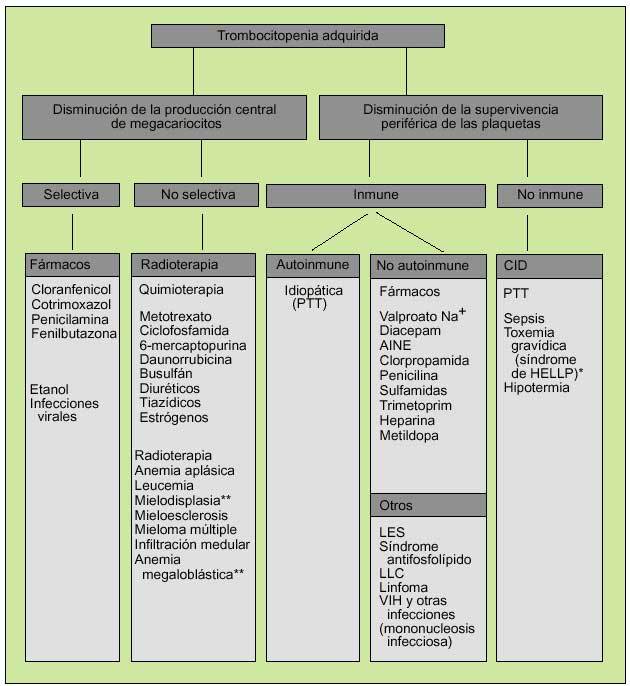
Fig. 5. Génesis de la trombocitopenia adquirida. *Síndrome de HELLP: anemia hemolítica, elevación de transaminasas y trombocitopenia que se produce en el tercer trimestre del embarazo. **Por trastornos de maduración.
Manifestaciones clínicas
Las manifestaciones clínicas de sangrado estarán en relación con el número de plaquetas o a su funcionalidad. Por encima de 50 por 109/l será rara la evidencia de sangrado. Por debajo de 10 por 109/l la incidencia de hemorragia espontánea será alta.
Son características las hemorragias mucocutáneas, en forma de petequias o púrpura, aunque también son habituales las epistaxis, las gingivorragias, las metrorragias y las hemorragias poscirugía. En casos graves, puede haber hemorragia digestiva, hematuria e incluso hemorragia subaracnoidea o hematoma subdural. Es excepcional la presencia de hemartros. Sin embargo, muchos pacientes con recuentos bajos (< 50 por 109/l) no presentan clínica de sangrado.
Exploración física y valoración clínica
Deberá incluir una minuciosa inspección en busca de evidencia de sangrado en sus diferentes manifestaciones cutáneas (recordar que si la púrpura es palpable apuntará a vasculitis y no a un trastorno de las plaquetas). Se palparán las distintas regiones linfáticas y se examinará de forma minuciosa hígado y bazo en busca de visceromegalias. La exploración clínica deberá ser completa, cara a descartar otro tipo de afección que secundariamente pudiera dar lugar a plaquetopenia: enfermedades del tejido conectivo, neoplasias, sepsis, etc., así como observar si el paciente está pálido y/o ha tenido fiebre o infecciones recurrentes, situaciones que pudieran apuntar a pancitopenia.
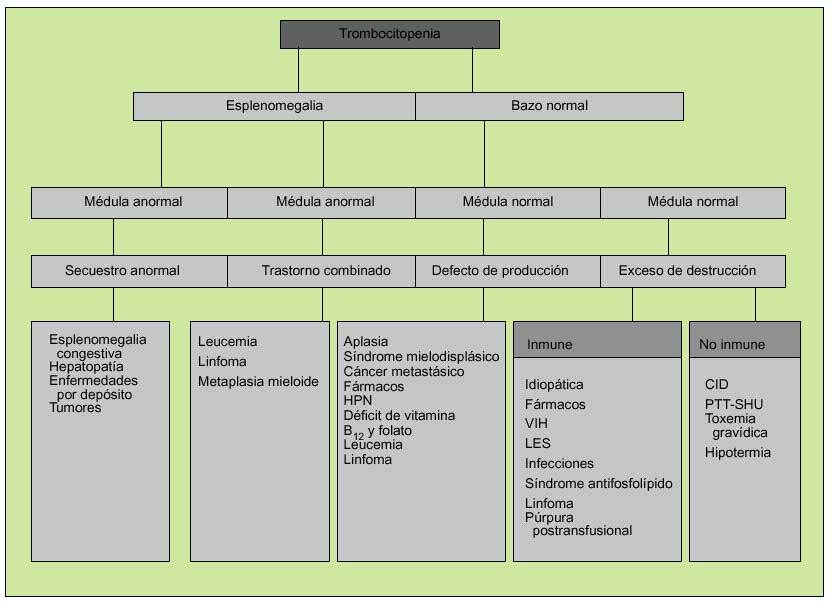
La aproximación clínica a la plaquetopenia variará según la presencia o ausencia de esplenomegalia y el estado de la médula ósea. Así, una esplenomegalia con un aspirado/biopsia medular normal apuntará hacia algún trastorno que dé lugar a secuestro esplénico, como cualquier causa de hipertensión portal, mientras que una esplenomegalia con examen medular anormal se deberá probablemente a leucemia o linfoma. En ausencia de esplenomegalia y médula ósea normal, la plaquetopenia se deberá a un exceso de destrucción periférica, cuyas causas podrán ser inmunes (TPI) o no inmune (CID) (fig. 6).
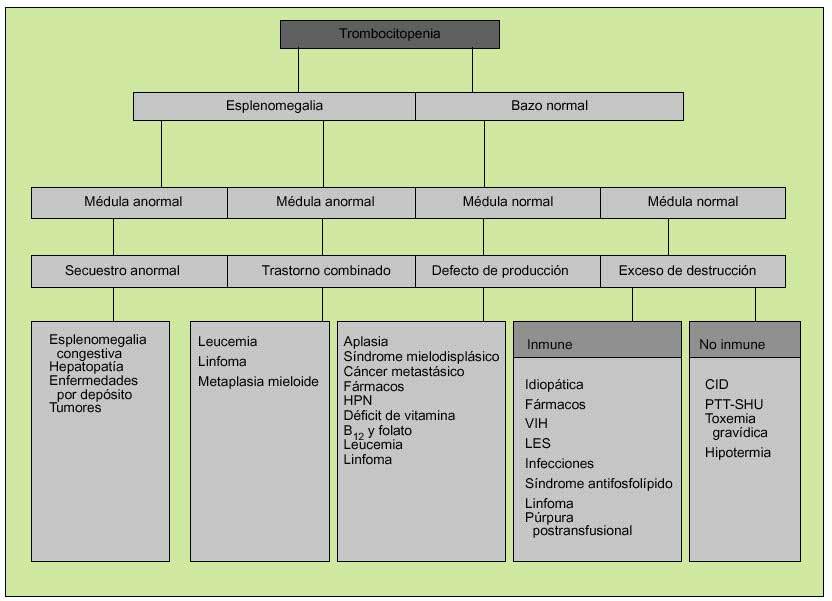
Fig. 6. Valoración clínica de la trombocitopenia (una vez descartada la pseudotrombopenia). HPN: hemoglobinuria paroxística nocturna; PTT-SHU: púrpura trombótica trombopénica-síndrome hemolítico-urémico.
Valoración diagnóstica
Se basará en una correcta anamnesis (excluir fármacos y enfermedades asociadas), exploración física y pruebas complementarias (laboratorio, aspirado-biopsia de médula ósea y pruebas de imagen).
Laboratorio
Hemograma. Se valorará el volumen plaquetario, que se encuentra elevado en las trombopenias congénitas y periféricas, y disminuido o de tamaño normal ante defecto de producción o secuestro esplénico. El hemograma nos informará, además, del estado del resto de líneas hematopoyéticas.
Estudio de coagulación. Se determinará el INR y PDF cuando se sospecha CID, y los anticuerpos antifosfolípidicos ante sospecha de dicho síndrome. Cuando el hemograma, el recuento de plaquetas y el examen del frotis sanguíneo son normales, se solicitará el tiempo de hemorragia para detectar déficit cualitativo de las plaquetas. Normalmente el sangrado cede a los
3-8 min.
En la mayoría de los pacientes con un tiempo de sangrado prolongado debido a plaquetas cualitativamente defectuosas, el defecto será adquirido; la causa más frecuente es el uso de ácido acetilsalicílico o derivados, seguido de la presencia de alguna enfermedad sistémica (uremia).
Bioquímica sérica. La función renal será útil ante sospecha de síndrome hemolítico-urémico.
Estudio inmunológico. Se solicitarán los anticuerpos antinucleares, ya que a veces la trombopenia es la primera manifestación de un trastorno autoinmune (LES).
La detección de anticuerpos antiplaquetarios es una prueba muy sensible pero poco específica de PTI, por lo que en la actualidad se considera una prueba innecesaria.
Serologías. Las serologías principales a solicitar son: virus de la inmunodeficiencia humana (VIH), virus de Epstein-Barr (VEB), citomegalovirus (CMV) y hepa-
titis.
Deberá realizarse un estudio de gota gruesa ante sospecha de malaria (la presencia de anemia y plaquetopenia en un paciente susceptible de malaria, debe hacer descartar esta enfermedad).
La aproximación diagnóstica al estudio de un trastorno plaquetario en un paciente con sangrado estará en función de si el recuento de plaquetas es normal o está disminuido, y se deben descartar en el primer grupo trastornos hereditarios y adquiridos del funcionalismo y en el segundo grupo, trastornos adquiridos, ya sean periféricos como centrales (tabla 2).

En aquellos pacientes con plaquetopenia, ausencia de ingestión de fármacos, megacariocitos en número normal o aumentado en ausencia de afección infiltrativa medular y de esplenomegalia, el diagnóstico más probable es el de PTI.
Pruebas de imagen
Se debe realizar una ecografía o la tomografía axial computarizada (TAC) abdominal ante hepato o esplenomegalia palpable o ante linfadenopatía generalizada.
Aspirado-biopsia de médula ósea
Nos dará el diagnóstico definitivo del origen de la trombopenia (central o periférica). Sin embargo, sólo estará indicada ante sospecha de causa central y nunca ante un diagnóstico favorable a PTI en el adulto. Asimismo, informará sobre otras causas primarias y secundarias como leucemia, infiltración metastásica, anemia megaloblástica, etc.
Púrpura trombocitopénica idiopática
Es un proceso autoinmune, generalmente idiopático, que cursa con destrucción de las plaquetas circulantes por la presencia de autoanticuerpos en contra de las proteínas de superficie, especialmente IIb-IIIa. La médula ósea no se encuentra afectada y no existe esplenomegalia ni adenopatías periféricas. Es relativamente común, con una incidencia más elevada en mujeres entre los 15 y 50 años, y es la causa más frecuente de plaquetopenia en ausencia de anemia o neutropenia. Puede asociarse a la presencia de otras enfermedades, especialmente del tejido conectivo (LES), infecciosas (HIV) o neoplásicas (linfoma y LLC).
Patogenia
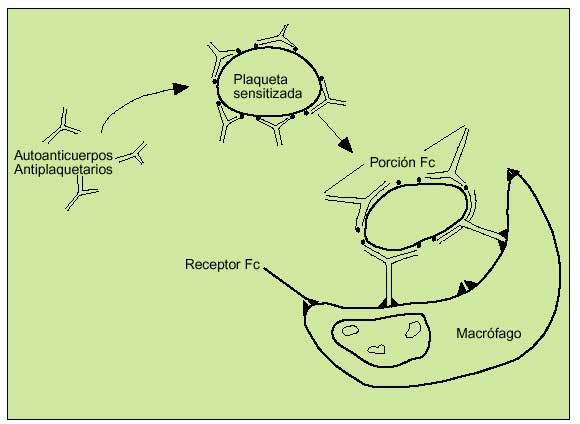
Las plaquetas sensibilizadas con autoanticuerpos dirigidos contra las glucoproteínas IIb-IIIa serán fagocitadas por los macrófagos en el bazo tras haberse unido a la fracción Fc del autoanticuerpo. Las plaquetas cubiertas por autoanticuerpos verán su vida de 7-10 días reducida a unas pocas horas. Los megacariocitos totales en médula ósea aumentarán de forma proporcional a la destrucción periférica (fig. 7).
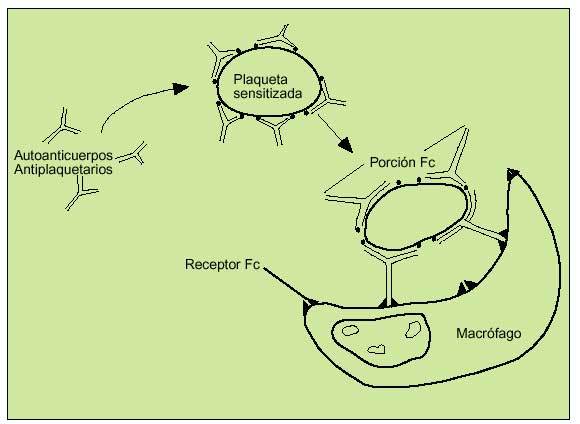
Fig. 7. Patogénesis de la PTI. Los macrófagos del sistema reticuloendotelial reconocerán la fracción Fc del autoanticuerpo que recubre a las plaquetas sensitizadas, que son destruidas por fagocitosis.
Formas evolutivas
Aguda
Es típicamente una forma infantil. Suele debutar tras infecciones del tracto respiratorio.
Se produce a través de la formación de complejos inmunes que cubren la superficie de la plaqueta y activan el complemento. También puede originarse por anticuerpos antivirales que presentan reacción cruzada con plaquetas, y activación del complemento.
Crónica
Es cuando su duración es superior a los 6 meses, siendo raro el desencadenante viral; es infrecuente aunque se ha observado un notable incremento en relación a la infección por el VIH. Se asocia también a otros trastornos autoinmunes, especialmente LES y anemia hemolítica Coombs positivo (síndrome de Evans). Ocurre con mayor frecuencia en mujeres entre la tercera y la cuarta década de la vida.
Mecanismo
Se produce por la formación de autoanticuerpos contra inmunoglobulinas plaquetarias de superficie, especialmente Gp IIb-IIIa. Las plaquetas serán destruidas por macrófagos tras la unión de éstos al fragmento Fc del complejo Ag/Ac (fig. 7).
Clínica
Inicio insidioso en forma de hemorragias petequiales, facilidad para la formación de hematomas y menorragias en mujeres. A veces pueden aparecer ampollas de contenido hemorrágico en la boca, que son características; epistaxis sólo en casos graves, y raramente hemorragia intracraneal. Sin embargo, la gravedad del sangrado en estos pacientes es menor que la que ocurre en trombopenias centrales con valores plaquetarios comparables, lo que se atribuye en la PTI a la circulación de formas más jóvenes, funcionalmente superiores. No existe esplenomegalia ni adenopatías a no ser que exista una enfermedad concomitante que lo justifique.
Diagnóstico
Será siempre por exclusión, en aquellas situaciones que se presenten con trombopenia aislada y ausencia de enfermedades sistémicas o exposición a fármacos y con una médula ósea normal o hipercelular.
El hemograma pondrá de manifiesto un número reducido de plaquetas, generalmente entre 10-50 por 109/l. Se solicitarán anticuerpos antinucleares para determinar la existencia de enfermedades concomitantes del tejido conectivo como el LES.
El aspirado de médula ósea revelará un número normal o ligeramente aumentado de megacariocitos.
Tratamiento
El tratamiento de urgencia se instaurará ante trombopenia asociada a sangrado y se llevará a cabo mediante prednisona a dosis de 1-2 mg/kg/día/vía oral. A las
3 semanas se efectuará un nuevo recuento y si éste es aceptable (cifras entre 80.000-90.000/µl3) se procederá a la retirada gradual. El 50% normalizarán sus cifras de plaquetas, aunque la mayoría sufrirá un descenso del número de plaquetas una vez dejen los corticoides.
Ante hemorragia importante, se emplearán, además de los corticoides, infusión de inmunoglobulinas a dosis de 400 mg/kg/día/5 días o 1 g/kg/día/3-2 días. El mecanismo de acción de esta terapia es mediante bloqueo de los receptores Fc en macrófagos.
Ante resistencia al tratamiento se deberá proceder a esplenectomía, respondiendo el 70-80% de forma parcial o completa, aunque en mayores de 45 años y mujeres posmenopáusicas se deberá intentar previamente el tratamiento con danazol (200-400 mg/12 h).
En aquellos pacientes refractarios a estas medidas se utilizarán inmunosupresores: ciclofosfamida en pauta continua (2 mg/kg/día) o en bolos (1 g/m2), azatioprina, ciclosporina A, dexametasona en bolos o interferón alfa. La transfusión de plaquetas sólo estará indicada ante compromiso vital.
Una mejoría espontánea tan sólo ocurrirá en un 10% de los pacientes con ITP crónica.
Bibliografía general
Cervera J, Sanz MA. Aproximación diagnóstica al paciente con trombocitopenia. Jano 1998; 1278: 63-68.
Handin RI, Robert I. Clotting dissorders. En: Harrison TR, editor. Principles of internal medicine (13.a ed). McGraw-Hill, 1992; 1798-1800.
Hoffbrand AV, Pettit JE. Platelets, blood coagulation and haemostasis. En: Essential haematology Londres: Blackwell Scientific Publications, 1994; 299-317.
Liesner RJ, Machin SJ. ABC d'Haematologia clínica: trastorns de les plaquetes. Ann Med 2000; 83: 262-266.
May M, Martín F. Leucopenia, trombopenia y pancitopenia. En: Muñoz B, Villa LF, editores. Manual de diagnóstico y terapéutica médica (Hospital Universitario 12 de octubre). Madrid: Díaz de Santos, 1993; 94.