









Parkinson's disease (PD) accounts for a significant burden on modern society, with an increasingly rapid growth in cases. Variants of several genes have been linked to the risk of developing the disease (e.g., the GBA gene). Other genes have been associated with autosomal dominant (e.g., SNCA, LRRK2, VPS35) and autosomal recessive PD (e.g., PRKN, PINK1, DJ-1). Such genes as 13A2, FBXO7, PLA2G6, SYNJ1, and DNAJC6 are associated with recessive forms displaying early onset, greater severity, and atypical characteristics. The genetic study of PD has clinical implications, shedding light on the underlying molecular mechanisms and potential therapeutic targets. This article presents a brief review of the molecular and genetic mechanisms and the phenotype–genotype relationship of GBA and other genes associated with autosomal dominant monogenic PD.
The prevalence of Parkinson disease (PD) has increased, representing a significant burden for modern society. Pathogenic variants in diverse genes have been related to the risk of PD (e.g., GBA). Specific pathogenic variants in several genes are causative of autosomal dominant PD (i.e., SNCA, LRRK2, VPS35) or autosomal recessive PD (i.e., PRKN, PINK1, DJ-1). Besides, genes like ATP13A2, FBXO7, PLA2G6, SYNJ1, and DNAJC6, are related to early-onset recessive parkinsonism, usually with a more severe progression and atypical features. PD genetics help in the understanding of the underlying molecular mechanism and the identification of potential therapeutic targets.
This article presents a brief overview of molecular mechanisms and genotypephenotype relation of autosomal-dominant and GBA-related PD.
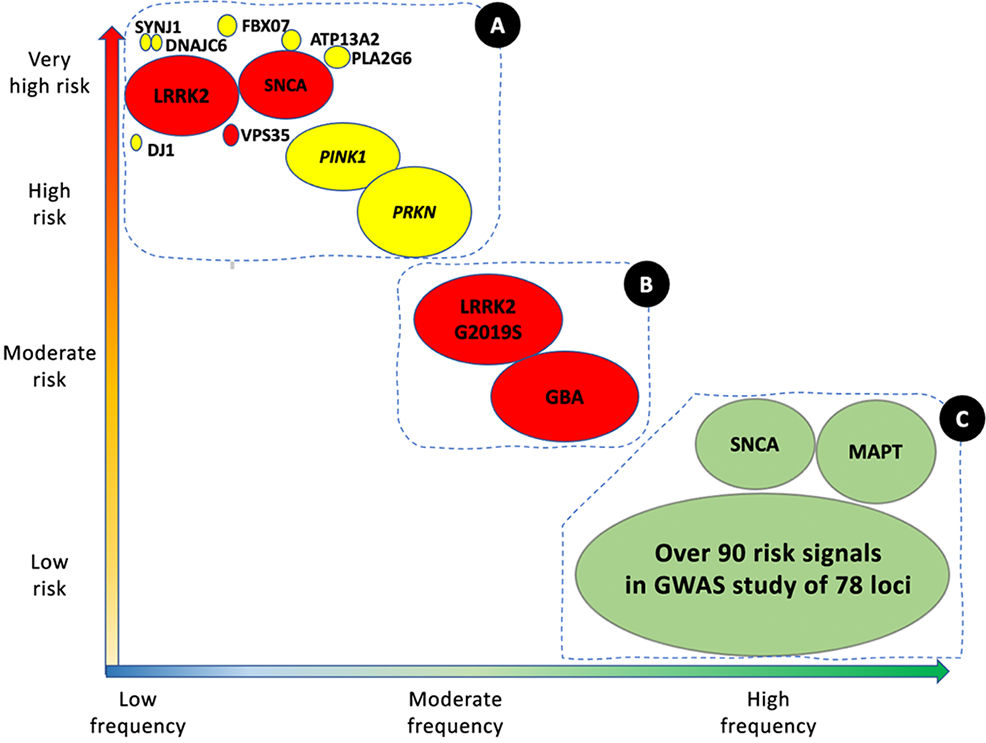
Parkinson's disease (PD) is the second most frequent neurodegenerative disease, after Alzheimer disease, and is an increasingly serious healthcare problem. With a prevalence of approximately 1% in the population aged over 65 years, and 4% among those older than 85 years, the total number of cases worldwide was estimated at 6.2 million in 2015, and is expected to grow to over 12 million by 2040.1 The majority of cases of PD are sporadic, of idiopathic cause.2 However, numerous genes have been shown to be involved in the disease. These can be categorised as: (1) rare variants with large effects (monogenic or familial PD), and (2) risk variants, with less pronounced effects, which are generally identified in sporadic cases (Fig. 1).

Genetic architecture of Parkinson's disease.
Continuum of genetic variants of different severity and frequency. The size of each bubble corresponds to the approximate population frequency of each allele. Colours indicate the form of inheritance: dominant (red), recessive (yellow), and risk loci (green). Genetic risk factors for PD can be divided into 3 groups.
Group A includes the majority of Mendelian genes associated with PD. These variants are associated with a significant risk of PD, but are very rare.
Group B includes more frequent variants that are associated with moderately high risk of PD. This group currently includes GBA1 and LRRK2 variants.
Group C includes variants associated with low risk of PD, but which are observed in a high percentage of patients with the disease.
This group includes over 90 potential independent genome-wide significant signals detected in a genome-wide association study meta-analysis of over 13 000 patients and 95 000 controls. Like LRRK2 and SNCA, some VPS13C and GCH-1 mutations are probably causal or associated with high risk of PD. Adapted from Schneider et al.75 (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Although 15% of patients with PD have family history of the disease, only 5%–10% of patients present a well-defined genetic cause.3,4 Monogenic cases of sporadic PD have also been reported (for instance, those related to SNCA mutations). The proportion of cases with genetic causes varies between cohorts; for instance, among patients undergoing implantation of deep brain stimulators, up to 29% carry variants of the PRKN, LRRK2, or GBA genes,5 whereas up to 40% of Arab-Berber patients and 20% of patients of Ashkenazi Jewish descent are carriers of the G2019S variant of LRRK2.6
To date, over 20 loci (PARK) have been associated with PD of Mendelian inheritance (Table 1), with SNCA, LRRK2, and VPS35 presenting autosomal dominant inheritance; more recently, cases have been associated with the genes EIF4G1, CHCHD2, and LPR10. In the autosomal recessive group, PRKN, PINK1, and DJ-1 express a similar phenotype to that of idiopathic PD,2,7–9 whereas ATP13A2, PLA2G6, FBXO7, DNAJC6, and VPS13C show atypical phenotypes.10,11 It has also been suggested that CHCHD2, LRP10, TMEM230, and UQCRC1 may be involved in PD of autosomal recessive inheritance. The list of candidate genes continues to grow, although in many cases their involvement is controversial due to lack of replication, functional studies, or pathological confirmation.10
Genes associated with monogenic Parkinson's disease.
| HGNC symbol (OMIM) | Full name | Locus/MIM code | Location | Inheritance | Early symptom onset | Certainty of association with PD | Lewy bodies |
|---|---|---|---|---|---|---|---|
| SNCA (163890) | α-Synuclein | PARK1 (168601) | 4T22.1 | Dominant | Yes* | Very high | Confirmed |
| PRKN (602544) | Parkin RBR E3 ubiquitin protein ligase | PARK2 (600116) | 6T26 | Recessive | Yes | Very high | Unconfirmed |
| PARK3 (602404) | PARK3 (unclear) | 2p13 | Dominant | Low | Unconfirmed | ||
| SNCA (163890) | α-Synuclein | PARK4 (168601) | 4T22.1 | Dominant | Yes | Very high | Confirmed |
| UCHL1 (191342) | Ubiquitin C-terminal hydrolase L1 | PARK5 (613643) | 4p13 | Dominant | Yes* | Low | Unconfirmed |
| PINK1 (608309) | PTEN-induced kinase 1 | PARK6 (605909) | 1p36 | Recessive | Yes | Very high | Unconfirmed |
| DJ1 (602533) | Protein deglycase DJ-1; parkinsonism-associated deglycase | PARK7 (606324) | 1p36.23 | Recessive | Yes | Very high | Unconfirmed |
| LRRK2 (609007) | Leucine-rich repeat kinase 2 | PARK8 (607060) | 12q12 | Dominant | Very high | Confirmed | |
| ATP13A2 (610513) | ATPase 13A2 | PARK9 (606693) | 1p36.13 | Recessive | Yes | Very high | Unconfirmed |
| PARK10 (?) | Parkinson disease 10 | PARK 10 (606852) | 1p32 | ? | Unconfirmed | Unconfirmed | |
| GIGYF2 (612003) | GRB10 interacting GYF protein 2 | PARK 11 (607688) | 2T37.1 | Dominant | Low | Unconfirmed | |
| PARK12 (?) | Parkinson disease 12 | PARK12 (300557) | Xq21-q25 | X-linked | Unconfirmed | Unconfirmed | |
| HTRA2 (606441) | HtrA serine peptidase 2 | PARK13 (610297) | 2p13.1 | Dominant | Yes* | Low | Unconfirmed |
| PLA2G6 (603604) | Phospholipase A2 group VI | PARK14 (612593) | 22q13.1 | Recessive | Yes | Very high | Unconfirmed |
| FBXO7 (605648) | F-box protein 7 | PARK15 (260300) | 22q12.3 | Recessive | Yes | Very high | Unconfirmed |
| PARK16 (?) | Parkinson disease 16 | PARK16 (613164) | 1T32 | ? | Unconfirmed | Unconfirmed | |
| VPS35 (601501) | VPS35 retromer complex component | PARK17 (614203) | 16q11.2 | Dominant | Very high | Unconfirmed | |
| EIF4G1 (600495) | Eukaryotic translation initiation factor 4 gamma 1 | PARK18 (614251) | 3T27.1 | Dominant | Low | Unconfirmed | |
| DNAJC6 (608375) | DnaJ heat shock protein family (Hsp40) member C6 | PARK19 (625528) | 1p31.3 | Recessive | Yes | High | Unconfirmed |
| SYNJ1 (604297) | Synaptojanin 1 | PARK20 (615530) | 21Q22.1 | Recessive | Yes | High | Unconfirmed |
| TMEM230 (617019) | Transmembrane protein 230 | PARK21 (616361) | 20p13 | Dominant | Yes* | Low | Confirmed |
| CHCHD2 (616244) | Coiled-coil-helix-coiled-coil-helix domain containing 2 | PARK22 (616710) | 7p11.2 | Dominant | Yes* | Low | Unconfirmed |
| VPS13C (608879) | Vacuolar protein sorting 13 homologue C | PARK23 (616840) | 15Q22.2 | Recessive | Yes | High | Unconfirmed |
| RIC3 (610509) | RIC3 acetylcholine receptor chaperone | 11p15.4 | Dominant | Yes* | Unconfirmed | Unconfirmed | |
| LPR10 (609921) | Low density lipoprotein receptor-related protein 10 | 14q11.2 | Dominant | Low | Confirmed |
* May also appear with early onset. HGNC: HUGO gene nomenclature committee. Source: https://www.omim.org/; Blauwendraat et al.12
Genome-wide association studies, which have mostly been conducted in European populations, have identified over 90 independent variants associated with the risk of developing sporadic PD.8,12 Key examples are variants of the GBA, SNCA, LRRK2, and MAPT genes.13
Recognising the role of genetics has epidemiological and pathophysiological implications. For instance, pathogenic variants of DJ-1, PRKN, PINK1, LRRK2, and SNCA affect mitochondrial function and mitophagy.15–17 Variants of GBA, LRRK2, and VPS35 are involved in lysosomal trafficking pathways (Fig. 2). Finally, establishing the genotype–phenotype relationship has implications for diagnosis and prognosis8; understanding these processes may enable the development of novel treatment strategies.14

Simplified depiction of the main molecular mechanisms associated with Parkinson's disease pathogenesis.
Oxidative stress, inflammatory response, mitochondrial dysfunction, and α-synuclein aggregation, together with impairment of the lysosomal and proteasomal degradation system, are the most significant PD-associated molecular mechanisms with a genetic basis.
ER: endoplasmic reticulum; TGN: trans-Golgi network.
This article presents a review of basic genetic aspects of PD, intended for clinicians interested in the disease.
ReviewGenetic risk factors in Parkinson's diseaseGBAThe GBA gene (1q21) encodes glucocerebrosidase (also known as glucosylceramidase), a lysosomal enzyme involved in sphingolipid metabolism. Its function is to catalyse the hydrolysis of the membrane lipid glucosylceramide into ceramide and glucose. Ceramide is a precursor of glycosphingolipids (e.g., GM1, GM2, and GM3 gangliosides) and sphingomyelin. Deficiency of this enzyme is associated with Gaucher disease, an autosomal recessive disease characterised by the accumulation of glucosylceramide in macrophages in visceral tissues, with a heterogeneous phenotype–genotype relationship.18,19
GBA variants are also strongly associated with synucleinopathies, considered the most important risk factor for the development of PD20; there is also evidence of heterozygous carriers among patients with Lewy body dementia.
The gene's role in PD involves several mechanisms:
- 1.
The gain-of-function hypothesis suggests a direct interaction between misfolded glucocerebrosidase and α-synuclein, increasing the accumulation and aggregation of the latter protein.21
- 2.
A loss-of-function hypothesis has been proposed, according to which structural changes reduce the activity of glucocerebrosidase due to failures in its transport across the endoplasmic reticulum or binding to its transporter. This decrease in activity would result in impaired sphingolipid homeostasis, affecting the trafficking, processing, and clearance of α-synuclein, with the consequent accumulation and aggregation of the protein.21
- 3.
A third hypothesis proposes a bilateral feedback loop in which glucocerebrosidase deficit facilitates α-synuclein oligomerisation, which in turn affects the activity of glucocerebrosidase, promoting a cycle of α-synuclein oligomer formation.22
- 4.
The lysosomal enzyme cathepsin D appears to mediate α-synuclein oligomer formation associated with GBA variants.23
The proportion of patients with PD carrying GBA variants varies as a function of ethnicity and the sequencing method used. The cumulative risk of developing PD among heterozygous carriers is 5% at 60 years of age, reaching 15%–30% by 80 years of age.4,20,24
The prevalence of GBA variants among patients with PD is estimated at 2.35%–9.4%, reaching 31.3% among patients of Ashkenazi Jewish descent.4 An Italian study using Sanger sequencing of the whole GBA gene in 874 patients with PD identified variants in 125 (14.3%), including 36 out of 176 (20.4%) with early onset PD and 89 out of 674 (13.2%) with late-onset PD. Of the 36 variants identified, N370S, L444P, and E326K were the most frequent, accounting for 24%, 23.2%, and 12.8% of the total, respectively.25
It should be noted that different pathogenic GBA variants present different levels of risk, and are associated with heterogeneous PD severity and progression.26 To date, over 500 variants of the gene have been reported (point mutations, insertions, deletions, frameshift mutations, splice site mutations, and recombinant alleles). These can be classified according to residual glucocerebrosidase activity (for instance, 13%–24% and 32%–38% for L444P and N370S, respectively) and the association with Gaucher disease (Table 2).25,26
Classification of GBA variants.
| Type of variant | Association | Examples |
|---|---|---|
| Mild | Non-neuronopathic Gaucher disease | N370S, R496H |
| Severe | Neuronopathic Gaucher disease | L444P, c.84insG, IVS2+1G->A, V394L, 370Rec |
| Risk | Associated with PDNot reported in Gaucher disease | E326K |
| Complex | 2 or more variants in cis due to the conversion, fusion, or insertion of parts of GBAP1 in GBAa | |
| Unknown | Unknown |
In general terms, patients with PD carrying pathogenic GBA variants present earlier onset than non-carriers,27 and more frequently present dystonia and shorter latency to onset of dyskinesia and motor fluctuations after onset of levodopa treatment.28 The akinetic-rigid phenotype is the most common form of onset,25 with gait freezing being more prevalent and more severe.29,30 Some studies report that cognitive impairment, visual hallucinations,31 anxiety, impulse control disorders, and non-motor fluctuations are also more frequent and appear earlier in carriers of severe variants.25
Parkinsonism onset is earlier in homozygous or compound heterozygous carriers than in heterozygous carriers of severe variants, and the latter present earlier PD onset than heterozygous carriers of mild variants.20,32
The risk of dementia in carriers of severe variants is 5.6 times greater than in idiopathic cases and 2.9 times greater than in carriers of mild variants.26
Comparison between carriers of mild and severe variants, adjusted for disease duration and medication, shows that the latter group scores higher in the Unified Parkinson's Disease Rating Scale and displays more advanced stages on the Hoehn and Yahr Scale.20 These patients also present more severe non-motor symptoms, such as depression, REM sleep behaviour disorder, anosmia, hallucinations, and shorter latency to onset of cognitive impairment, compared to those with milder variants and idiopathic cases.25
Patients carrying GBA variant E326K, one of the most frequent variants, present fast progression of motor symptoms, postural alterations, and greater risk of cognitive impairment.20,26
Genes associated with Parkinson's disease of autosomal dominant inheritancePARK-SNCAFunction of the α-synuclein proteinThe SNCA gene encodes α-synuclein, a presynaptic protein of 140 amino acids that is involved in the compartmentalisation, storage, and recycling of neurotransmitters including dopamine. It also promotes membrane curvature, acts as a chaperone to the synaptic SNARE complex, and contributes to synaptic vesicle trafficking and dopamine release.33,34 The proteins potentially interacting with α-synuclein include chaperones/chaperonins, translocation factors, enzymes, mitochondrial-associated proteins, and proteins involved in secretion and degradation.35 It has been suggested that α-synuclein may modulate DNA repair.36 There is also evidence that it modulates intramitochondrial Ca2+ signalling, participates in mitochondrial fusion/fission, and is involved in electron transport chain complexes and axonal transport.37 Furthermore, its expression in immune cells and haematopoietic cells suggests a functional role in these processes.38
Normal structure and pathological changesIn its native state, α-synuclein occurs as a monomer, or as an α-helical homotetramer. The latter structure comprises 2 α-helices followed by an acidic C-terminal region, which is fundamental to interaction with other proteins: the first α-helix is positively charged and can bind to lipid membranes, whereas the second contains the non–amyloid-beta component, a hydrophobic region that enables the protein to form beta-sheets (Fig. 3A).33,35 The N-terminal acetylation or extension, comprising 10 amino acids, also results in tetramer formation, even without interaction with membrane phospholipids. The current paradigm suggests a dynamic equilibrium between these 2 conformational states.34
 Amino acid sequence of SNCA; double helically folded tetrameric conformation of α-synuclein and the main PD-associated SNCA variants at the N-terminal domain. B) Different conformational states of α-synuclein and its aggregation in fibrils and Lewy bodies. Source: Fig. 3A taken from Post et al.76")
SNCA gene: molecular structure, genetic variants, and pathological forms of α-synuclein.
A) Amino acid sequence of SNCA; double helically folded tetrameric conformation of α-synuclein and the main PD-associated SNCA variants at the N-terminal domain. B) Different conformational states of α-synuclein and its aggregation in fibrils and Lewy bodies. Source: Fig. 3A taken from Post et al.76
The accumulation in neurons of α-synuclein aggregates is believed to be a key process in PD pathogenesis. Patients present high levels of α-synuclein, with detergent-insoluble inclusions of the protein. The largest and most organised forms of these aggregates are incorporated in Lewy bodies, a pathological marker of the disease, which contain phosphorylated and ubiquitinated forms of α-synuclein (Fig. 3B).39 Accumulation mechanisms include defects in the proteasome degradation pathway, which may be reciprocally inhibited by the presence of oligomeric forms or fibrillary species of α-synuclein.40 The pathological role of α-synuclein in its different conformational states has been a subject of debate in recent decades. Some theories suggest that toxicity is mediated by excessive levels of abnormal α-synuclein (Fig. 4), or by loss of function of the protein in its native state.41
, defects of inter-organelle contact (blue box), and dysfunction of organelle dynamics (orange box). ER: endoplasmic reticulum. Adapted from Wong and Krainc.77 (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)")
Pathways involved in the α-synuclein toxicity theory.
α-Synuclein toxicity has been linked to organelle dysfunction (yellow boxes), defects of inter-organelle contact (blue box), and dysfunction of organelle dynamics (orange box).
ER: endoplasmic reticulum.
Adapted from Wong and Krainc.77 (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
SNCA variants cause autosomal dominant parkinsonism with aggregation of α-synuclein, possibly due to accelerated fibrillation or oligomerisation, and progressive loss of nigrostriatal dopaminergic neurons.42SNCA A53T, described in 1997, was the first genetic variant associated with familial cases of PD,42 and is the most frequent point mutation, particularly in Italian and Greek families; it has also been reported in sporadic cases. Other point mutations include A30P (in German families), E46K (in Basque families), and more recently H50Q, G51D, and A53E.33 The different variants appear to be heterogeneous in terms of phenotype, pathological behaviour, and functional effects on α-synuclein (Table 3). Though rare, a growing number of cases of gene copy number variations (duplications and triplications) of SNCA have been reported.
Comparison of the effects of different SNCA variants, compared with the native-state gene, on age of onset, lipid binding, and fibril formation.
| Variant | Age of onset | Lipid binding | Rate of α-synuclein fibre formation |
|---|---|---|---|
| A53T | 20–85 | ↑ | ↑ |
| E46K | 50–69 | ↑ | ↑ |
| H50Q | 60–71 | ≅ | ↑ |
| A30P | 54–76 | ↓ | ↓ |
| G51D | 19–61 | ↓ | ↓ |
| A53E | 36 | ↓ | ↓ |
Table reproduced with permission from Meade et al.34
SNCA-PD onset generally occurs in the fifth decade of life (<1% before 20 years of age and approximately 33% between 20 and 40 years and 66% after 40 years of age).43SNCA “burden” is correlated with age of onset, with a mean age of onset of 46.9 (10.5) years in patients with duplications and 34.5 (7.4) in those with triplications.44
According to data from literature reviews, over 70% of patients present rigidity and bradykinesia and 30%–80% present resting tremor43,45; the latter symptom is less frequent in patients with the SNCA A53T variant.46 Postural instability is observed in 89% of cases, and 83% present atypical signs (e.g., anterocollis/retrocollis, pyramidal signs, alien hand syndrome).43
Compared to idiopathic cases, SNCA-PD more frequently presents with dementia, hallucinations, and depression.45 Carriers of the A53T or E46K variants more frequently present dementia than carriers of the A30P variant, which is associated with milder phenotypes.34 56% of patients with multiplications report dementia in the first 5 years after onset of motor symptoms. Dementia onset is estimated to occur at 59 years and 42 years in carriers of duplications and triplications, respectively.44,47,48
Levodopa is usually effective,49 with up to 78% of patients responding well,43 although its effect may be short-lived. Among responders, 44% developed dyskinesia and/or motor fluctuations, and 24% presented dystonia.49
Psychotic symptoms were reported in 80%–95% of carriers of triplications and duplications, with heterogeneous rates reported in patients with point mutations (20%–76.2%).43,45,50
Depression is reported in approximately 22% of patients with SNCA-PD due to point mutations, 40%–56% of those with duplications, and 55%–100% of those with triplications.43,45
Autonomic signs are also frequent, occurring in 60% of patients with triplications, 40% with duplications, and 48%–56% of patients with the A53T variant.43,45 Some patients present severe orthostatic hypotension.46
Only weak evidence is currently available on other SNCA polymorphisms; a meta-analysis has associated some of these with increased risk of PD, including the rs2736990, rs356220, rs356165, rs181489, rs356219, rs11931074, and rs2737029 variants51; rs356219 is associated with a smaller effect on disease progression,52 whereas rs356195 may increase the risk of impulsive behaviour.53 In general, no association has been shown between the known SNCA polymorphisms and clinical heterogeneity of PD.
PARK-LRRK2Function of the LRRK2 proteinLRRK2 is a multi-domain protein belonging to the ROCO family. It includes domains regulating protein–protein interactions, such as the N-terminal armadillo repeat, ankyrin repeat, leucine-rich repeat, and the C-terminal WD40 domain. It also presents 2 enzymatic domains, the Ras of complex (ROC) GTPase domain and the serine/threonine kinase domain, separated by a C-terminal ROC domain. Familial mutations tend to affect catalytic domains.
Various functions have been attributed to LRRK2 due to its phosphorylation, via the kinase/GTPase domains, of over 20 substrates; these functions include cytoskeleton remodelling, protein expression, synaptic transmission, and membrane trafficking, among others.54
LRRK2 pathways associated with PDG2019S, the LRRK2 variant most frequently associated with familial PD, is associated with greater levels of kinase activity than in the wild-type protein. Other variants, such as R1441C/G/H and Y1699C, show increased binding to GTP, reduced GTP hydrolysis, and greater kinase activity. On the contrary, loss-of-function variants have not been found to increase the risk of PD.55
Cell and animal models have shown excess LRRK2 protein to be neurotoxic due to kinase-dependent mechanisms. Dysfunction of these pathways would facilitate the neurodegenerative process. In addition to the pathological detection of Lewy bodies in patients with LRRK2-PD, there is evidence that the protein is involved in pathogenesis through its molecular and functional relationship with α-synuclein.54 The identification of multiple Rab GTPases, G-proteins that regulate cellular vesicle trafficking, as physiological substrates of LRRK2 has given rise to an interesting hypothesis linking increased LRRK2 activity to cell-to-cell transmission of α-synuclein.54
LRRK2 variantsPathogenic variants of LRRK2 are risk factors for familial and sporadic (“idiopathic”) PD. LRRK2-PD is considered the most frequent form of monogenic autosomal dominant parkinsonism, presenting incomplete penetrance (age-dependent). Carriers of these variants account for 6%–40% of familial cases, depending on the ethnic group, and up to 2% of sporadic cases.54
A founder effect explains the accumulation of specific variants in certain populations, such as the G2019S variant in Ashkenazi Jew and Arab-Berber populations, with a south–north gradient in European countries; this is the most frequent variant worldwide. R1441G is the most frequent variant in the Basque Country, and displays a north–south gradient in Spain. Rarer pathogenic variants include Y1699C (reported in Germany, Denmark, Canada, and the United Kingdom), R1441C (Nebraska), I2020T (Japan), N1437H (Norway and Sweden), and R1441H.56 The G2385R variant has been identified as a risk factor in Japanese and Korean populations; in China, it is reported in 8%–11.7% of patients with PD and 0.5%–3.3% of the general population.57
LRRK2-PD phenotypeThe penetrance of G2019S is estimated at 25%–100% at 80 years of age,57 whereas the mean age of onset of LRRK2-PD is estimated at 57 years. Only 6% of patients present early onset.43
Generally, 88% of patients report tremor at some point during disease progression; similar figures are reported in carriers of the R1441G (76%) and G2019S variants (89%).43 Tremor is the initial symptom in 63% of patients with G2019S LRRK2-PD and 52% of idiopathic cases.58
In a cohort of patients of Ashkenazi Jewish descent with early-onset G2019S LRRK2-PD, a greater tendency to the postural instability/gait alteration phenotype was observed,59 with greater prevalence of falls compared to non-carriers.60
Dystonia, including “off” painful dystonia, is reported in 38%–42% of patients with LRRK2-PD, compared to approximately 25% of those with idiopathic PD.43,58
G2019S LRRK2-PD presents approximately 3 times greater prevalence of dystonia than idiopathic PD in the first 2 years, although, unlike in recessive parkinsonism, this rarely occurs before onset of dopaminergic treatment.
Atypical signs are rarely reported in LRRK2-PD (9%)43; as in idiopathic cases, 95% of patients respond well to levodopa,43,49 with 64% and 68% presenting dyskinesia and motor fluctuations, respectively.43 The latter symptom is more frequent in carriers of the R1441C/G/H variants than in those with other variants.43,61
Cognitive impairment and dementia appear to be less frequent in LRRK2-PD,47 only occurring in approximately 23% of patients,43,45 with dementia prevalence below the reported rate of 11%.48 Carriers of the G2019S variant present better attention, executive function, and language than non-carriers, after adjusting for phenotype.62
Psychotic symptoms are reported in 7%–32% of patients,43,45 whereas data on depression in G2019S carriers are controversial,63 with both higher and lower levels being reported than those seen in idiopathic PD.59,64,65
Patients with G2019S LRRK2-PD present higher rates of insomnia and lower rates of REM sleep behaviour disorder than in idiopathic cases.64 In a report of 18 cases of LRRK2-PD, 3 presented sleep attacks and 6 presented excessive daytime sleepiness.66
Autonomic signs are estimated to present in approximately 22.5% of patients with LRRK2-PD,45 with G2019S carriers presenting normal R-R interval variability67 and greater cardiac MIBG uptake; orthostatic hypotension is rarely reported.68
It has been suggested that patients with G2019S LRRK2-PD present greater risk of cancer than non-carriers, including skin cancer, leukaemia, breast cancer in women, and prostate cancer in men.69
PARK-VPS35Function of the VPS35 proteinVPS35 encodes vacuolar protein sorting ortholog 35 (VPS35), a subunit of the retromer complex, the main actor in the endosomal network (endoplasmic reticulum, Golgi apparatus, and endosomes); it is located in the early endosome and controls retrograde trafficking of cargo proteins from the early endosome to the trans-Golgi network or plasma membrane. The retromer complex also mediates vesicular transport from mitochondria to peroxisomes. This complex is the main centre that receives, dissociates, and sorts cargoes from different sources: (a) the plasma membrane (recycling of membrane receptors), (b) biosynthetic pathways (recovery of trafficking from the Golgi apparatus), and (c) the lysosomal pathway (direct trafficking to lysosomes). Generally, these activities control the homeostasis of transmembrane proteins at the plasma and endoplasmic levels, and regulate the abundance of receptors, signalling receptors, adhesion molecules, and hydrolase receptors.70
VPS35 pathways associated with PDThe retromer complex plays a role in neurodegenerative processes through its control of synaptic function and the trafficking and location of cargo proteins essential in neuronal homeostasis and in the balance between α-synuclein accumulation and clearance. VPS35 variants may cause PD through interference with α-synuclein degradation pathways.70
VPS35 variantsThe prevalence of the D620N variant is estimated at approximately 1.3% in familial cases of PD and 0.3% in sporadic cases.71
VPS35-PD phenotypeVPS35-PD presents a mean age of onset of 50 (7.3) years,43,72 with early onset in only 7% of cases.43 Most patients respond well to levodopa. In a group of 8 responders, 4 reported dyskinesia, 5 presented motor fluctuations, and 2 displayed dystonia.43 Tremor is reported in 77%–98% of cases, and postural instability in 60%–67%, with no atypical signs.43,72
A review reported cognitive impairment in 44.5% of patients undergoing evaluation; however, after inclusion of patients for whom these data were missing, only 6% showed cognitive impairment.45 Psychotic symptoms appear to be less frequent (2/8 patients; 25%),43,45,63 whereas depression was reported in 10/15 patients (66.6%).45 Most of these patients present excessive daytime sleepiness,63 and a minority (4.5%) present autonomic signs.45
ConclusionsOnly a minority of cases of PD are of monogenic cause, although polymorphisms are a frequent genetic risk factor. Study of genetic mechanisms enables understanding of pathophysiology, definition of prognostic implications, and the creation of new treatment strategies.
Heterozygosity for GBA mutations is the most significant genetic risk factor identified to date. Severe mutations of the gene are associated with high risk of early-onset dementia; this demonstrates the importance of establishing phenotype–genotype relationships. Treatments seeking to modulate glucocerebrosidase enzyme activity and levels of α-synuclein in the central nervous system may modulate the progression of PD.73
Despite their low prevalence, research into SNCA mutations may shed light on the functioning of α-synuclein and the possible effects of toxicity or loss of function on its folding and pathological aggregation. An interesting aspect is the inverse “dose-dependent” effect of duplications and triplications on age of PD onset, which supports the theory that α-synuclein “burden” may play a key role in PD pathophysiology.
LRRK2 mutations account for the majority of autosomal dominant familial PD, with a phenotype indistinguishable from that of idiopathic PD. Besides their frequency, the importance of these mutations lies in their potential association with certain cancers, which may lead to the development of screening strategies, and the therapeutic potential of LRRK2 inhibition as a disease-modifying drug in future.74
To continue the advance towards precision medicine, it is essential to improve our understanding of the genetics of PD, the molecular pathways involved, and phenotype–genotype relationships.
Supplementary material.
