



Chediak–Higashi syndrome (CHS) is a rare and potentially fatal autosomal recessive disease characterized by frequent bacterial infections, bleeding tendency, oculocutaneous albinism, photosensitivity and progressive neurologic dysfunction. Owing to the rarity of this condition, the objective of this study was to describe patients with CHS.
MethodsRetrospective evaluation of patients followed in a paediatric tertiary centre of Allergy and Immunology of São Paulo, Brazil, between 1986 and 2018 with a confirmed diagnosis of CHS. Data were obtained from medical records. Demographic aspects, family history, clinical findings, laboratory data, diagnosis, treatment and outcome were described.
ResultsA total of 14 patients (five male) were included. Clinical manifestations were first recognized at a median age of two months (at birth-20 months). Median age at diagnosis was 1.7 years (0–5 years). All patients had recurrent infections. Albinism was present in 13 patients and silvery or light hair was present in 14. Seven patients developed hemophagocytic lymphohistiocytosis (HLH); the median age at the diagnosis of HLH was 5.7 years (2.6–6.7 years) and the median interval between the diagnosis of CHS and HLH was 3.3 years (0–5 years). Four of the most recently diagnosed patients underwent bone marrow transplantation (BMT). Nine patients are deceased, and one was lost to follow-up. The median age of death was 6.7 years (3.8–22 years). Five patients died of HLH, one of lymphoma, and three of infection. All the patients who had HLH before the year of 2000 died of HLH. The two most recently diagnosed patients with HLH were able to cure the HLH, although they died of other causes. Four patients are alive, three of them after successful BMT.
ConclusionThirty years of follow up showed an improvement in the prognosis in patients with CHS. The better understanding of the underlying biological mechanisms of HLH allowed the standardization of management protocols, resulting in survival improvement. BMT is the only treatment that can change CHS prognosis, which emphasizes the need for early identification of the disease.
Chediak–Higashi syndrome (CHS) is a rare autosomal recessive disease with fewer than 500 cases reported worldwide.1,2 CHS was first described in 1943 and is characterized by frequent bacterial infections, bleeding tendency, oculocutaneous albinism, photosensitivity, and progressive neurologic dysfunction.3
A phenotype-genotype correlation has been suggested, involving mutations in the LYST gene.2,4 The main cellular characteristic of CHS is the accumulation of giant organelles (lysosomes/granules) in different cells including neutrophils, lymphocytes, melanocytes, and platelets due to abnormal vesicle trafficking.1,2,5 The CHS immunological features are characterized by normal or reduced number of natural killer cells with reduced function, neutropenia and impaired neutrophil function.2 The classic form, described in 85% of patients, progresses to an accelerated phase, currently named hemophagocytic lymphohistiocytosis (HLH). HLH involves excessive lymphocyte and macrophage activation and is the most frequent cause of death in CHS, typically in the first or second decade of life.6 The atypical form has a milder course and does not progress to HLH.
The diagnosis is established by the identification of giant granules within neutrophils in a peripheral blood smear and/or by mutations in LYST on molecular genetic testing.2,7
The only treatment is bone marrow transplant (BMT), which corrects the immune and hematologic defects, but does not prevent the progressive neurologic dysfunction.6,8
Despite the syndrome’s classic and pathognomonic features, diagnosis can be delayed because of its rarity. A prompt diagnosis is essential to improve the outcome of the disease since infections can be prevented, HLH can be more easily suspected and treatment with BMT can be offered to patients with CSH.
Owing to the rarity of the disease, the recent recognition of CHS as a cause of primary HLH and its indication for BMT, our group proposed to describe the patients diagnosed with CHS over three decades. This study allowed us to analyse the natural history of the disease as well as the impact of BMT on the disease course.
MethodsRetrospective evaluation of patients followed in a paediatric tertiary centre of Allergy and Immunology of São Paulo, Brazil, between 1986 and 2018 with a confirmed diagnosis of CHS. Data were obtained from medical records. Demographic aspects, family history, clinical findings, laboratory data, diagnosis, treatment, and outcome were described.
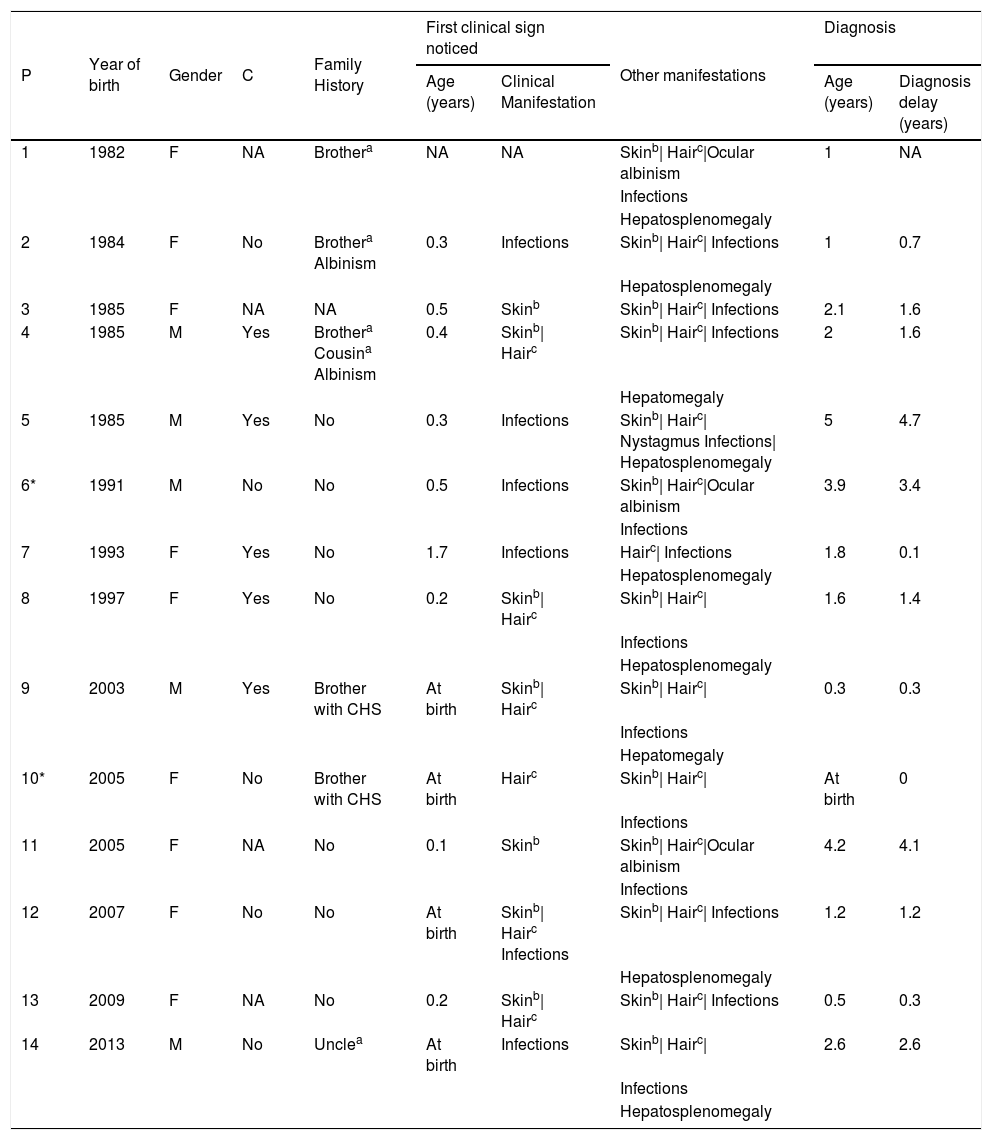
ResultsDemographic and clinical characteristics of 14 patients with CHS are summarized in Table 1. The median age at diagnosis ranged from 0 to 5 years old, with a median age of 1.7 years old; five were male. Five children had consanguineous parents and six had a positive family history: a relative with premature death (4/6), phenotypic similarity (4/6) or CHS diagnosis (2/6).
Demographic and clinical data.
| P | Year of birth | Gender | C | Family History | First clinical sign noticed | Other manifestations | Diagnosis | ||
|---|---|---|---|---|---|---|---|---|---|
| Age (years) | Clinical Manifestation | Age (years) | Diagnosis delay (years) | ||||||
| 1 | 1982 | F | NA | Brothera | NA | NA | Skinb| Hairc|Ocular albinism | 1 | NA |
| Infections | |||||||||
| Hepatosplenomegaly | |||||||||
| 2 | 1984 | F | No | Brothera Albinism | 0.3 | Infections | Skinb| Hairc| Infections | 1 | 0.7 |
| Hepatosplenomegaly | |||||||||
| 3 | 1985 | F | NA | NA | 0.5 | Skinb | Skinb| Hairc| Infections | 2.1 | 1.6 |
| 4 | 1985 | M | Yes | Brothera Cousina Albinism | 0.4 | Skinb| Hairc | Skinb| Hairc| Infections | 2 | 1.6 |
| Hepatomegaly | |||||||||
| 5 | 1985 | M | Yes | No | 0.3 | Infections | Skinb| Hairc| Nystagmus Infections| Hepatosplenomegaly | 5 | 4.7 |
| 6* | 1991 | M | No | No | 0.5 | Infections | Skinb| Hairc|Ocular albinism | 3.9 | 3.4 |
| Infections | |||||||||
| 7 | 1993 | F | Yes | No | 1.7 | Infections | Hairc| Infections | 1.8 | 0.1 |
| Hepatosplenomegaly | |||||||||
| 8 | 1997 | F | Yes | No | 0.2 | Skinb| Hairc | Skinb| Hairc| | 1.6 | 1.4 |
| Infections | |||||||||
| Hepatosplenomegaly | |||||||||
| 9 | 2003 | M | Yes | Brother with CHS | At birth | Skinb| Hairc | Skinb| Hairc| | 0.3 | 0.3 |
| Infections | |||||||||
| Hepatomegaly | |||||||||
| 10* | 2005 | F | No | Brother with CHS | At birth | Hairc | Skinb| Hairc| | At birth | 0 |
| Infections | |||||||||
| 11 | 2005 | F | NA | No | 0.1 | Skinb | Skinb| Hairc|Ocular albinism | 4.2 | 4.1 |
| Infections | |||||||||
| 12 | 2007 | F | No | No | At birth | Skinb| Hairc Infections | Skinb| Hairc| Infections | 1.2 | 1.2 |
| Hepatosplenomegaly | |||||||||
| 13 | 2009 | F | NA | No | 0.2 | Skinb| Hairc | Skinb| Hairc| Infections | 0.5 | 0.3 |
| 14 | 2013 | M | No | Unclea | At birth | Infections | Skinb| Hairc| | 2.6 | 2.6 |
| Infections | |||||||||
| Hepatosplenomegaly | |||||||||
Siblings P: patient; F: female; M: male; C: consanguinity; NA: not available.
Clinical manifestations were recognized at a median age of two months (at birth-20 months). Phenotypic signs were the first features to be noticed in seven patients: partial albinism with heterogeneous skin pigmentation and/or hair coloration change. One of those children was born with dark hair and the albinism and hair colour change were progressive. In five cases, recurrent infections were the first warning signs to be detected and in one partial albinism with recurrent infections were observed at the same time.
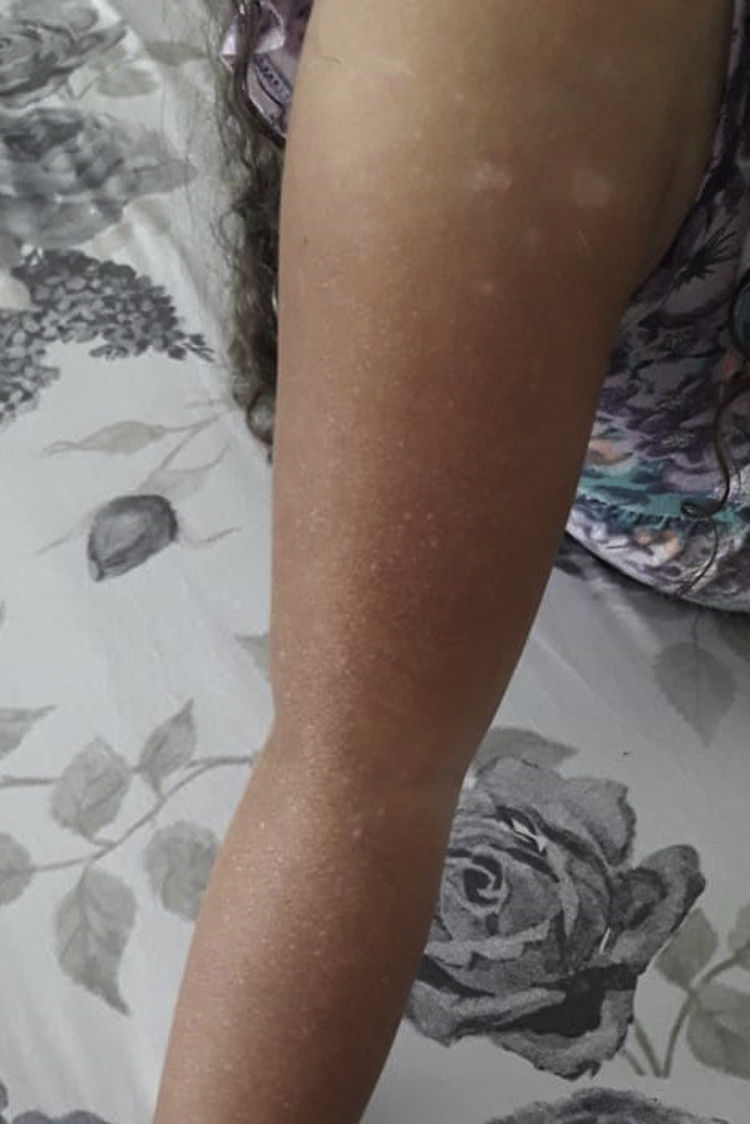
At physical exam on admission, 13 children had silvery hair and one had light hair; 13 had partial albinism that ranged from hyperpigmented skin face to hyper and hypopigmented generalised macules (Figs. 1 and 2). One had a normal skin exam. Ocular albinism was detected in four patients: three had decreased retinal pigmentation on ocular examination (3/3) and one had nystagmus. Information regarding ocular evaluation was not available for the remaining eleven patients. Seven patients had hepatosplenomegaly and two had isolated hepatomegaly.


Laboratory findings at diagnosis were: anaemia in 12, neutropenia in ten, leukopenia in four and thrombocytopenia in four patients. All the patients had giant granules in peripheral blood smear, and nine (9/9) also had giant granules in bone marrow aspirate. Microscopic analysis of the hair was done in four patients and revealed rounded and evenly distributed melanin granules. Genetic tests were not performed in any of the cases.
The median interval between first clinical manifestation and diagnosis was 1.4 years (0–4.8 years).
All children had recurrent infections in the course of the disease, including pneumonia (11/14), skin infections (9/14), otitis media (9/14), tonsillitis (7/14), diarrhoea (8/14) and sepsis (2/14). One patient had an adverse reaction to the Bacillus Calmette–Guerin vaccine (axillary adenopathy). There were no bone or central nervous system infections.
Seven patients developed HLH. Median age of HLH presentation was 5.7 years old (2.6–6.7 years) and the median interval between the diagnosis of CHS and HLH was 3.3 years (0–5 years). In one patient (P14) CHS was diagnosed after HLH and a simultaneous infection with Epstein-Barr virus (EBV). During HLH, two children developed neurologic manifestations: one patient (P5) presented seizures after central nervous system bleeding, and another patient (P7) had status epilepticus, dystonia, pyramidal signs and cognitive deterioration. These manifestations were part of the HLH neurologic impairment that culminated in the death of the two patients. Four patients had bleeding diatheses. HLH management was heterogeneous and different classes of medications were used: corticotherapy (6/7), immunosuppressive therapy (3/7), chemotherapy (3/7), intravenous immunoglobulin (2/7) and splenic radiotherapy (1/7). Additional medications were used during HLH event: antibiotic therapy (5/7), antiviral and antifungal therapy (2/7), folic acid (1/7), anticonvulsant therapy (1/7) and granulocyte-macrophage colony stimulating factor (1/7).
One patient (P12) developed acute myeloid leukaemia (AML) after being treated with dexamethasone and etoposide for HLH. Another patient (P6), who presented a milder form of the disease, developed diffuse large B cell lymphoma. Two patients (P6 and P8) manifested neurologic impairment, one had cerebellar ataxia and the other had mild cognitive impairment and cerebellar ataxia.
Four patients underwent bone marrow transplantation (BMT); two from unrelated and two from related donors. One patient (P14) had HLH before BMT.
Nine patients are deceased, and one was lost in follow-up. The median age of death was 6.7 years old (3.8–22 years). Five patients died of HLH, one of lymphoma, one of septic shock during the chemotherapy for AML, one of septic shock two months after BMT and one of infection. The median time between HLH diagnosis and death was three months (1–20 months). The other four patients are alive, three of them after successful BMT with a follow-up of 21 months to eight years (P10, P11, P13) and one is now a 20.3-year-old female with a mild clinical phenotype (P8). This patient now has a mild cognitive impairment and cerebellar ataxia, and still has recurrent infections, mostly skin infections with abscess.
DiscussionCHS is a rare and heterogeneous disease, with few case series described in the literature.8–12 We report one of the largest single-centre case series of CHS. The identification of 14 patients over a period of 30 years in a reference centre of a large country like Brazil, even considering the rarity of this disorder, could be related to the disease underdiagnosis. From the 14 patients in this case series, seven were previously described in another report from our group 20 years ago.7 The retrospective study of cases over three decades allows a reflective analysis about the evolution in the recognition and management of this disorder.
Recurrent infections are a major sign of primary immunodeficiencies and are also an important feature of CHS. In our series, all the patients had recurrent infections. The prevalence as well as the severity of infections described in other reports are variable.2,9
A positive family history for an affected child or consanguinity, although not always present, are important clues that should be actively inquired about when evaluating a child with recurrent infections.2 In one of our cases, the previous diagnosis of an affected brother allowed the diagnosis of CHS at birth, and the patient was submitted to BMT before the development of severe infections and HLH, prolonging the patient’s survival.
The oculocutaneous albinism should also be considered a red flag in a child with recurrent infections. However, skin albinism in CHS is highly variable and sometimes can only be noticed when comparing patients with their relatives’ phenotype.2 In this case series, skin albinism was not perceived in five patients. Interestingly, in one patient, the albinism became progressively more evident as the child grew up, and another patient did not have cutaneous manifestations despite having silvery hair, as previously described in other case reports.9 This variability also contributes to diagnosis delay. Considering this, a high level of suspicion is necessary, and a careful phenotype assessment is mandatory when evaluating children with severe infections or even with HLH.
An ophthalmologic evaluation must be included in the initial approach of these patients to screen for ocular albinism. The follow-up should be performed at least once a year, since ocular albinism can cause decreased retinal pigmentation, nystagmus, and diminished visual acuity.2
Differential diagnosis must include some rare disorders that present with silvery hair or albinism. The silvery hair syndromes (Griscelli and Elejalde syndromes) are characterized by silvery hair, pigment abnormalities, and central nervous system dysfunction. They differ from CHS as the characteristic leukocytic granules are absent in the other syndromes. Hair microscopy can also contribute to the differential diagnosis: in CHS it shows evenly distributed melanin aggregates of regular diameter and in Griscelli syndrome the melanin granules are large and mostly found in the medullary zone. Elejalde syndrome has a normal immunological function and hair microscopy reveals small and large clumps of melanin in an irregular pattern.2 Hermansky Pudlak syndromes should also be considered in the differential diagnosis since they are associated with oculocutaneous albinism, platelet storage pool deficiency and, in some subtypes, immunodeficiency.2
Peripheral blood smear confirms the diagnosis by the identification of pathognomonic giant granules in neutrophils, but they may be overlooked in an automated haematology analyser. The presence of giant cytoplasmic inclusions in myeloblasts or myeloid precursors resembling those seen in the CHS (pseudo-Chediak–Higashi granules) are described in acute myeloid leukaemia, and less commonly in chronic myeloid leukaemia and myelodysplastic syndromes and a few other disorders.2
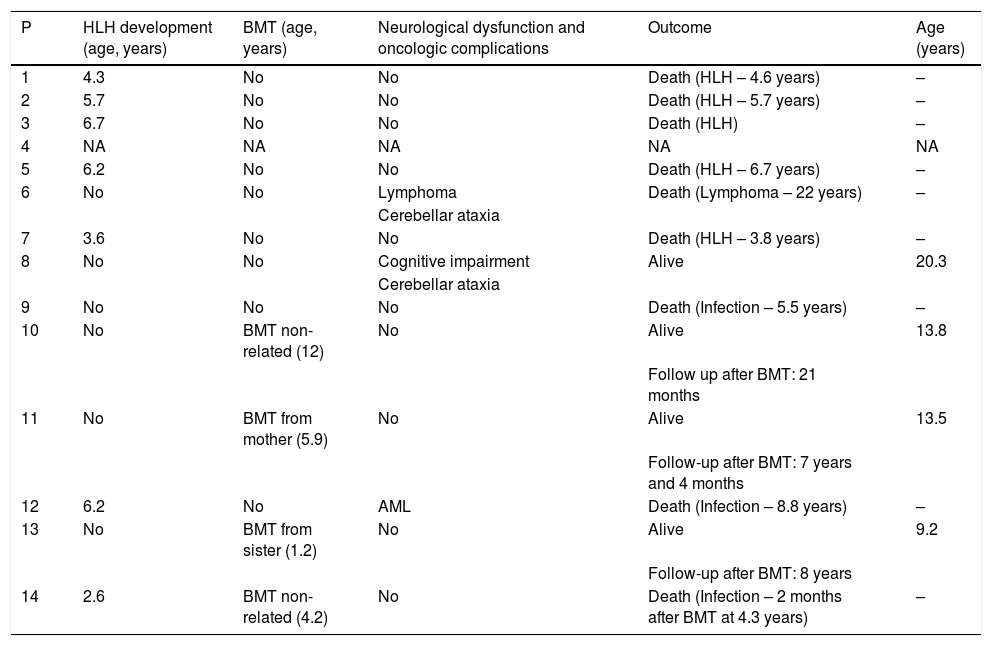
HLH is the most serious complication of CHS. It is an exacerbated inflammatory response, which is considered one of the 12 warning signs for immunodeficiencies in the first year of life.13 In fact, HLH can be the first presentation of CHS, as observed in one of our patients who had HLH triggered by EBV.11 Infections are important triggers of HLH (most frequently viral infections as EBV), even in the primary genetically determined HLH.14 In our series, half of the patients developed HLH, which is below the 85% incidence described in the literature, closer to the 33% incidence of the Japanese report.9 Despite the small number of cases in this series, we can observe an improvement in the recognition and management of CHS and HLH in our patients in the last three decades (Table 2), although HLH is still an important cause of death in patients with CHS. The first prospective international treatment protocol for HLH was introduced in 1994, and in the last decades the crescent understanding of the underlying biological mechanisms of HLH has enabled the standardization of therapeutic and management protocols, resulting in an improvement in survival.15 Considering the patients of this case series, all the patients that had HLH before the year of 2000 died of HLH. The two more recently diagnosed patients with HLH were cured of HLH and one of them survived until BMT, although they died of other complications.
Clinical evolution and outcome.
| P | HLH development (age, years) | BMT (age, years) | Neurological dysfunction and oncologic complications | Outcome | Age (years) |
|---|---|---|---|---|---|
| 1 | 4.3 | No | No | Death (HLH – 4.6 years) | – |
| 2 | 5.7 | No | No | Death (HLH – 5.7 years) | – |
| 3 | 6.7 | No | No | Death (HLH) | – |
| 4 | NA | NA | NA | NA | NA |
| 5 | 6.2 | No | No | Death (HLH – 6.7 years) | – |
| 6 | No | No | Lymphoma | Death (Lymphoma – 22 years) | – |
| Cerebellar ataxia | |||||
| 7 | 3.6 | No | No | Death (HLH – 3.8 years) | – |
| 8 | No | No | Cognitive impairment | Alive | 20.3 |
| Cerebellar ataxia | |||||
| 9 | No | No | No | Death (Infection – 5.5 years) | – |
| 10 | No | BMT non-related (12) | No | Alive | 13.8 |
| Follow up after BMT: 21 months | |||||
| 11 | No | BMT from mother (5.9) | No | Alive | 13.5 |
| Follow-up after BMT: 7 years and 4 months | |||||
| 12 | 6.2 | No | AML | Death (Infection – 8.8 years) | – |
| 13 | No | BMT from sister (1.2) | No | Alive | 9.2 |
| Follow-up after BMT: 8 years | |||||
| 14 | 2.6 | BMT non-related (4.2) | No | Death (Infection – 2 months after BMT at 4.3 years) | – |
P: patient; HLH: hemophagocytic lymphohistiocytosis; NA: not available; AML: acute myeloid leukaemia. BMT: bone marrow transplantation.
BMT is the only treatment that corrects the hematopoietic defects and can prevent HLH.15 It should be performed before HLH develops, but the ideal timing is unknown, giving the heterogeneity of the disease.2,9 In a multicentric review of 35 cases with CHS who underwent hematopoietic stem cell transplantation, the five-year probability of overall survival was 62%.8 In our centre, TMO only became available for the CHS patients after the year of 2010. Of the four patients who underwent BMT in our series, one died from a BMT complication but the other three are still alive.
Two patients of this series (P6 and P8) had a milder disease course. They were not submitted to BMT and did not have HLH. These cases are probably related to the 15% of patients described in the literature that have the atypical and milder form of the disease, which can progress to adulthood, although with neurologic sequelae.2,4,8
Neurologic disease, both central and peripheral, may take two or three decades to develop.6 However, in a three-case report of cord blood transplantation in children with CHS, they all had some neurologic sequelae at the age of six, eight and 15 years old.12 In the Japanese series, 4/15 patients presented neurologic manifestations and Umeda et al. described 2/8 patients with neurologic disease.9,10 In our series neurologic disease was identified in two patients (cognitive delay and tremor). A neurologic evaluation should always be performed and cognitive impairment, peripheral neuropathy, ataxia, and parkinsonism should be recognized as signs of disease progression even after BMT.2,6,8
Two children that were not submitted to BMT developed oncologic disease (lymphoma and AML). Oncologic disease was described in one patient in the Japanese series, and a case of myelodysplasia was described in another patient, but is not classically described in CHS.8,9
CHS is a rare disease and a cause of primary HLH. The presence of recurrent infections in a child with the typical phenotype should raise suspicion, especially in the presence of positive family history. The presence of giant cytoplasmic granules in neutrophils and lymphocytes in peripheral blood are diagnostic and should not be missed. CHS is potentially fatal but BMT can improve CHS prognosis. This emphasizes the need for early identification of affected children.
Conflict of interestNone.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
