



Respiratory diseases are currently the most common disorders seen in paediatric clinical practice. Although infections are the most frequent underlying causes, structural alterations may also be responsible. Congenital malformations of the lung are rare disorders occurring with variable degree of severity. Pulmonary artery agenesis is an infrequent congenital anomaly embryologically resulting from early involution of the proximal portion of the sixth aortic arch during development of the heart.1 The condition may manifest with total or partial absence of the lung on the same side (estimated incidence of total pulmonary agenesis is 0.0034–0.0097%2), with the presence of a hyperplastic and compensating contralateral lung.
Bilateral pulmonary artery agenesis is exceptional and incompatible with life. In such situations the right pulmonary branch generally manifests as an isolated lesion, while the left pulmonary branch is usually associated to cardiovascular malformations,1 with an increased incidence among males.
Pulmonary artery agenesis is usually well tolerated and manifests with repeated infections, haemoptysis and pleuritic chest pain, among other symptoms. Tolerance becomes complicated if the condition is moreover associated to vascular compression, bronchomalacia, tracheal stenosis, or other malformations.3
The occurrence of asthma in patients with congenital lung anomalies is extremely rare. This association has been reported only twice before, both of whom were adults when documented with pulmonary agenesis and associated asthma. The paucity of such a report in children in the literature prompted this description of an 8-year-old boy with pulmonary aplasia who also had asthma.4
In children, the affected side of the chest is usually decreased in size, with a reduction in inspiratory amplitude, scoliosis, diminished vesicular breath sounds and dullness in response to percussion on the affected side. Radiologically, displacement is observed of the mediastinum, heart and large vessels towards the affected hemithorax, with a reduction in intercostal space and elevation of the diaphragm. Diagnostic confirmation is based on magnetic resonance imaging (MRI), while establishing the absence or hypoplasia of the pulmonary artery requires an angiographic MRI (angio-MRI) or angiographic computed tomography (angio-CT) exploration. These techniques offer better sensitivity and specificity than echocardiography and arterial catheterisation.2
We show an 8-year-old male who reported to the Emergency Service with fever (peak 39.5°C), cough and mucus production for the previous three days. He had suffered two isolated vomiting episodes with mucus content, and partial rejection of food ingestion. This patient had a diagnosis of allergic asthma and allergic rhinoconjunctivitis in response to dust mites. Maintenance treatment had been provided with desloratadine in solution and montelukast chewable tablets (5mg) once a day for the past two years. He had had occasional wheezing and dyspnoeic episodes, particularly in response to physical exertion, with adequate response to inhalatory salbutamol.
Family history: Maternal uncle with histiocytosis X.
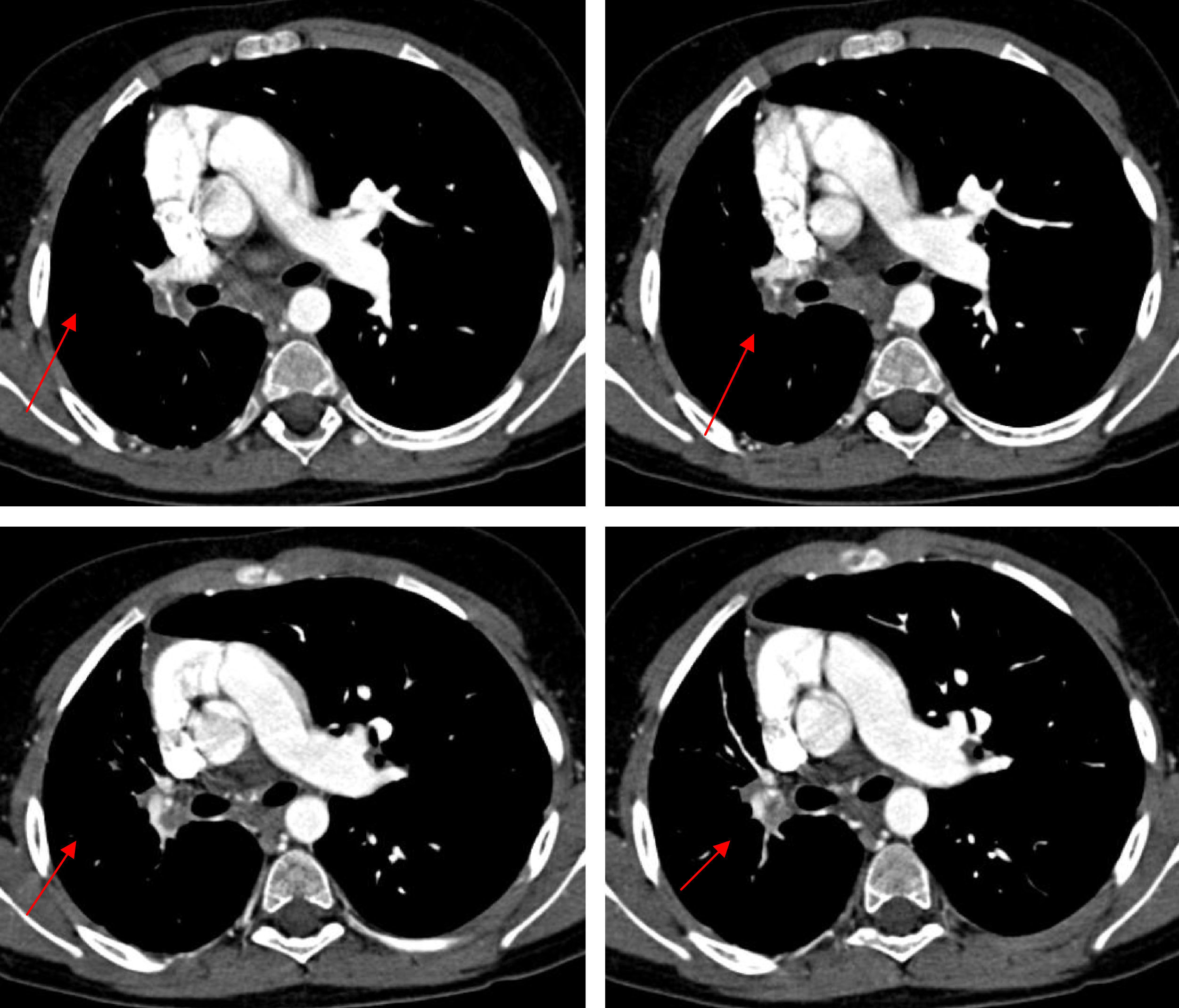
Personal history: Gestational age: 40 weeks. Normal prenatal ultrasound findings. Amniocentesis: normal. Karyotype: 46XY. Induced labour, vaginal delivery without instrumentation. Apgar 9/10. Weight 3.895kg (P75). Height 54.5cm (>P97). Cranial circumference: 34cm (P). No amniotic fluid pathology. The patient was admitted 16h after delivery due to respiratory difficulties, with a diagnosis of pneumomediastinum, pulmonary hypertension and minimal interatrial communication. He was also admitted at 8 months of age due to bronchopneumonia. The chest X-rays revealed bilateral basal infiltration, with an interstitial pattern. Posteriorly, he experienced several repeat pneumonia episodes in the right hemithorax, which were resolved with oral amoxicillin (80mg/kg/day during 10 days). The clinical examination revealed pale skin, while chest auscultation identified right-side hypoventilation, with basal subcrepitants. There was no intercostal indrawing or signs of breathing difficulty. Weight 40kg (P97), height 137cm (P97), temperature 37.9°C, SatO2 98%, heart rate 62bpm, respiratory frequency 28rpm, blood pressure 118/89mmHg, and no other relevant findings. Complementary explorations: Tuberculin test: negative. Pharyngeal smear: negative. Inspiration chest X-ray (Figure 1A): asymmetric lung volume with smaller size right hemithorax. The expiration X-ray (Figure 1B) in turn showed persistence of the asymmetry, with no air entrapment. Chest angio-CT without contrast injection (Figure 2): absence of right pulmonary artery with diminished volume of the entire right lung, thickening of the interlobular septae, and presence of enlarged-diameter bronchial, phrenic and intercostal arteries in the entire affected hemithorax. No tracheobronchial tree or mediastinal alterations were noted. Visceral-atrial situs solitus. Doppler ultrasound: Normal atrioventricular and ventriculoarterial communications. Intact intracardiac septae. No significant valve disease. Normal global ventricular and segmental function. No ductus or aortic coarctation. Right pulmonary artery agenesis. The spirometry tests revealed an obstructive pattern with a diminished forced vital capacity (1.1) (80%) and reduced FEV1 (1.52 (70%). Significant reversal (210ml) was noted after salbutamol inhalation. The findings therefore allowed us to establish a diagnosis of right pulmonary artery agenesis.
 Figure 2.
Figure 2.Consecutive angio-CT images with parenchymal window, showing absence of the right pulmonary artery with diminished volume of the entire right lung, thickening of the interlobular septae, and presence of enlarged-diameter bronchial, phrenic and intercostal arteries in the entire affected hemithorax.
(0.47MB).Since 1673 there have been reports indistinctly of both pulmonary agenesis and hypoplasia – mostly in newborn and young nursing infants.
The diagnosis is particularly difficult to establish when the anatomy is markedly distorted. However, CT and MRI allow us to assess the adjacent airway structures and determine the magnitude of vascular compression, tracheal stenosis and tracheal ring fusion, etc., with the added possibility of facilitating surgical planning if necessary.2
The right pulmonary artery is reportedly more often affected, since the contralateral side of the aortic arch is normally implicated. A left side location is more often associated with cardiovascular malformations, although this was not confirmed in our case.5
The literature documents some cases showing irrigation of the parenchyma on the affected side through the persistence of segmental arteries, collateral circulation, intercostal arteries, etc. This is explained by the different embryological origin of these structures, and supports the need for angio-MRI or CT with contrast injection for full evaluation of the patient.6 The above explains our case, in which right pulmonary artery agenesis was associated to the partial presence of ipsilateral pulmonary tissue. The Spencer classification catalogues the patients as:
 and expiration (B), showing asymmetric lung volume with smaller size right hemithorax. No air entrapment is seen.")

- •
type I (bilateral complete agenesis).
- •
type II (unilateral pulmonary agenesis): subtype a (associated total absence of bronchi), subtype b (presence of a rudimentary bronchus without lung tissue), and subtype c (poorly differentiated lung parenchyma, the latter corresponding to our own patient).
- •
type III (lobar agenesis).
Close to 30% of all affected patients may remain asymptomatic for life. However, pulmonary agenesis is associated with chronic and progressive inadequate lung function secondary to the poor development of the tissues, associating skeletal alterations and recurrent respiratory infections. These patients may develop pulmonary hypertension and can present other associated congenital anomalies. Follow-up of the patient and the evaluation of a possible familial or genetic aggregation is important.7
Of particular importance for the diagnosis of our patient was the history of repeated respiratory infections. Detailed evaluation of this phenomenon is needed in the context of pulmonary artery agenesis. Respiratory infections are common in childhood, the prevalences being 4%, 2% and 1% per year in children aged 5, 5–9, and over 9 years, respectively. Of these children, a small proportion develop recurrent or persistent respiratory symptoms, as in our patient. The classification of the anatomical distribution of the lungs may be of help in establishing the aetiology of recurrent or chronic pneumonia. The possible causes of disseminated or diffuse infiltrations may be the existence of metabolic, immune or neurological anomalies, while recurrent involvement of a single lung, lobe or segment is suggestive of airway or parenchymal malformations, or the result of atelectasis or hyperinsufflation secondary to obstruction of an airway segment.8
Cardiac or large vessel anomalies can give rise to recurrent local pulmonary infections via three mechanisms: 1) diminished blood supply secondary to deficient ventilation (possible explanation in the case of our patient); 2) venous circulatory alterations; or 3) extrinsic large vessel compression of the airway.8
The patient prognosis depends on the age at onset of the symptoms, the location of the disorder, the associated malformations, and the presence of lesions on the contralateral side.
Fallot tetralogy is described as the most common cardiac malformation associated to pulmonary artery agenesis — a condition that can be ruled out in our case in view of the age and symptoms of the patient, and the diagnostic exploratory findings.5 Other related anomalies have also been reported, such as hemivertebras, hemifacial microsomia, tracheal anomalies, renal alterations or genitourinary malformations, gastrointestinal, hepatic, thyroid or neurological malformations, etc.9
At present, the proposed management is based on symptomatic treatment, together with surgery in selected cases. It is estimated that 19–25% of all patients with pulmonary artery agenesis have pulmonary hypertension in adulthood. Knowing that pulmonary hypertension is characterised by a poor production of nitric oxide, possible pharmacological treatment has been evaluated in the form of sildenafil (a selective inhibitor of the enzyme 5-phosphodiesterase, which degrades nitric oxide), with a view to reducing pulmonary hypertension secondary to pulmonary artery agenesis.5
Considering that our patient showed no symptoms, no treatment was decided. Two years after the diagnosis the boy continues to lead a normal life.
The present case underlines the need for a high level of suspicion to diagnose congenital lung anomalies, and shows that asthma may appear in such individuals. In this context, appropriate asthma treatment can alleviate the symptoms and reduce morbidity.
