




In 2019, the World Health Organization (WHO) estimated that around 296 million people in the world were chronically infected with HBV and could transmit HBV to others [1]. Chronic Hepatitis B (CHB) occurred in 5 - 10% of adults infected, 25–50% of children under the age of five, and 90% of prenatal infections [2–5]. About 25% of CHB patients may develop liver cirrhosis or hepatocellular carcinoma (HCC), leading to high mortality [6,7].
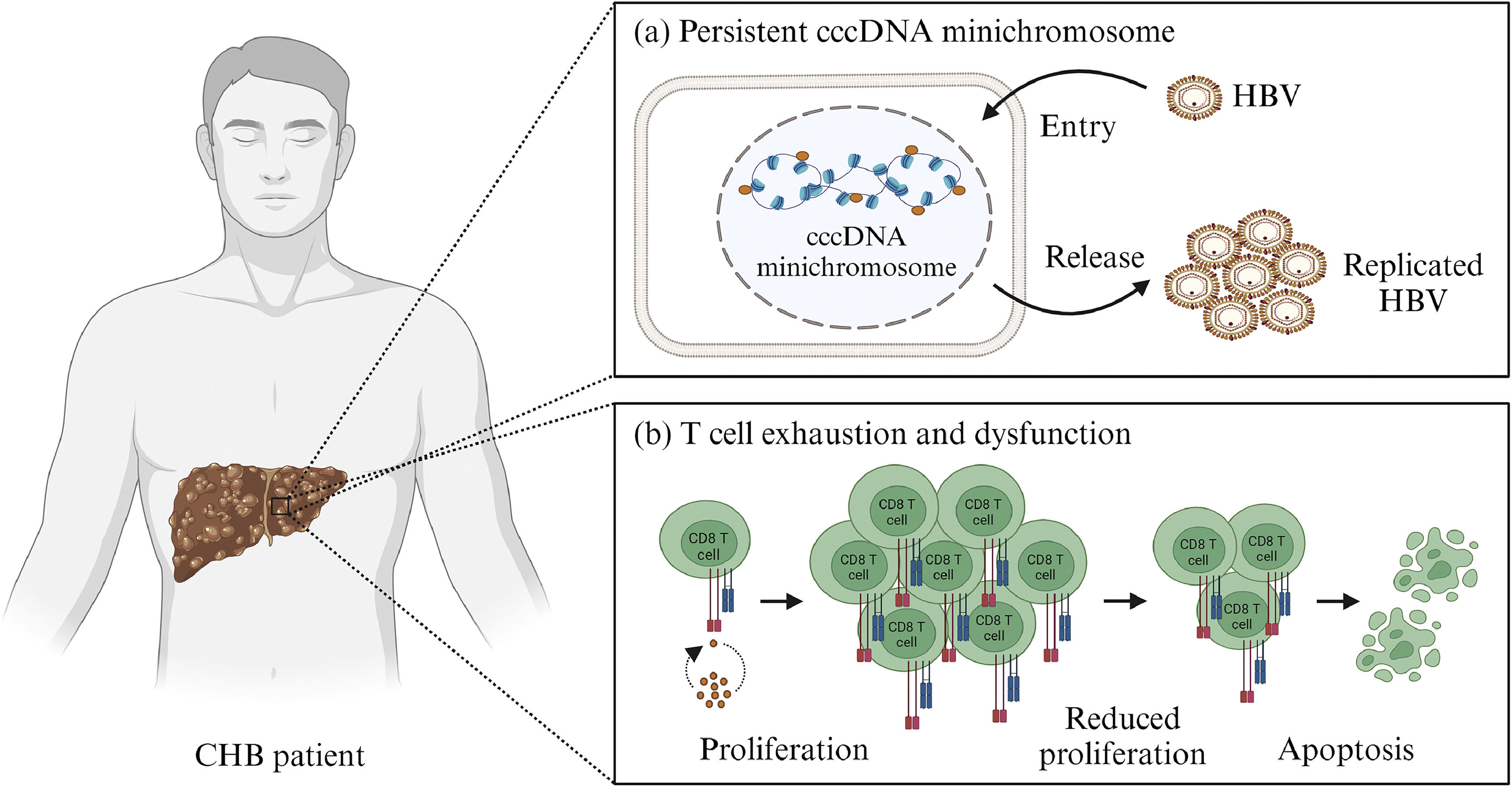
CHB has at least two major issues, including the persistent presence of HBV covalently closed circular DNA (cccDNA) in hepatocytes [8–11] and immune system disorders [12–15] (Fig. 1). cccDNA was discovered in varying levels in the hepatocytes of CHB patients, ranging from 1 to 15 copies per cell [8,9,16–20]. In hepatocytes, cccDNA associates with histone and non-histone proteins to form minichromosome. Epigenetic regulatory mechanisms such as DNA methylation, histone modifications including acetylation, ubiquitination, phosphorylation, small ubiquitin-related modifier (SUMO)-ylation, ADP ribosylation and deamination, chromatin remodeling, as well as interactions with non-coding RNA all affect the stability, conformation, and transcription of the cccDNA minichromosome [8–11]. This regulatory process involves several host cell proteins and also two HBV proteins, HBc and HBx [8–11,21,22]. Several factors contribute to immune system disorders in CHB patients, including the number of HBV, antigen exposure, and an unsupportive liver metabolic environment [12–15,23]. CHB patients are known to have exhaustion and dysfunction in both T and B cells, as indicated by a reduction in the number of these cells [12,14,24]. This could explain why the immune system of CHB patients fails to eliminate the HBV cccDNA minichromosome.
. (a) The presence of Hepatitis B virus (HBV) covalently closed circular DNA (cccDNA) minichromosome allows HBV replication to continue in liver cells. (b) Prolonged antigen exposure can cause exhaustion and dysfunction in T cells, one of which is defined by a reduction in T cell number due to apoptosis. Created with BioRender.com.")
Main problems with chronic hepatitis B virus infection (CHB). (a) The presence of Hepatitis B virus (HBV) covalently closed circular DNA (cccDNA) minichromosome allows HBV replication to continue in liver cells. (b) Prolonged antigen exposure can cause exhaustion and dysfunction in T cells, one of which is defined by a reduction in T cell number due to apoptosis. Created with BioRender.com.
Currently, there are two types of antiviral therapy used to treat CHB, pegylated interferon alpha 2a (PEG-IFNα) and nucleos(t)ide analogues (NA) [6,25,26]. PEG-IFNα interacts with the IFNα receptor complex to modulate the transcription of several IFN-stimulated genes [9,27]. Unfortunately, PEG-IFNα has several adverse effects, including the production of thyroid autoantibodies, cytopenia, and neuropsychiatric symptoms such as depression and insomnia [6,26,28]. Furthermore, the effectiveness of this treatment varies according to HBV genotype. CHB patients with HBV genotype A have better treatment outcomes than other genotypes [25,29–31]. NA can inhibit the activity of DNA polymerase [32]. However, long-term usage of first-generation NAs such as Lamivudine and Adefovir can lead to resistance [33,34], while second-generation NAs like Telbivudine may cause renal toxicity [6]. NA therapy, however, is unable to eliminate cccDNA; therefore, it does not accomplish sterilizing cure [8], the functional cure rate is only 1–12% [26,28,35], and discontinuing NA use may lead to relapse in patients [36].
Therapeutic vaccines have the potential to treat CHB and are considered superior to antiviral therapy because they can exploit the ability of dendritic cells to present antigens and improve the response of CD8 T cells to eliminate HBV-infected hepatocytes [37]. For therapeutic vaccine development, there are several things need to be considered, including the natural properties of the antigen [13]. The antigen selected as the main component of the CHB therapeutic vaccine must induce both innate and adaptive immune responses, especially CD8 T cells, which have a cellular response [38]. Naturally, antigen expression should not be excessive to avoid exhaustion and dysfunction of T cells [24]. This antigen must have a role in the epigenetic regulation of cccDNA to ensure the subsequent immune response not only reduces HBV levels but also eliminates or inhibits cccDNA transcription. This paper aims to explore HBV antigens suitable for epigenetic manipulation of cccDNA, elucidate their mechanisms of action, and evaluates their potential as key components of epigenetically-driven vaccines for CHB therapy.
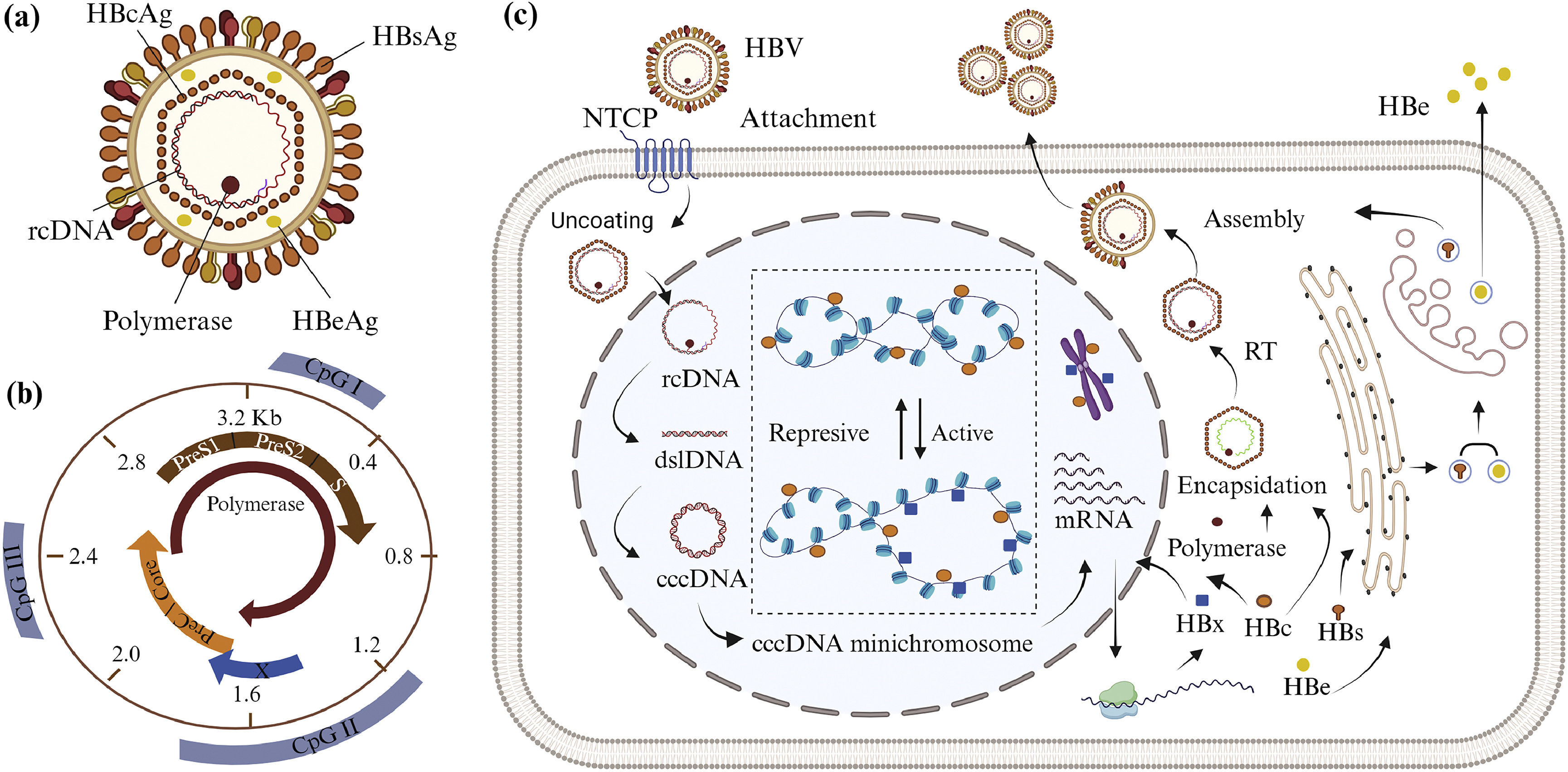
2HBV life cycle and minichromosome cccDNA formationHBV is the smallest DNA virus in the Hepadnaviridae family. The virion has a double-layered envelope and an icosahedral nucleocapsid (Fig. 2A). It is classified as class VII in the Baltimore classification because the genetic material is in the form of relaxed circular DNA (rcDNA), which replicates via intermediary RNA using the reverse transcriptase enzyme [39]. The HBV life cycle begins with the virion binding to the surface of the host cell (Fig. 2C). The virion envelope protein binds to hepatocyte receptors, sodium-taurocholate cotransporting polypeptide (NTCP), and the virions enter the cell by endocytosis [22,40,41]. The nucleocapsid releases HBV genetic material in the form of 3.2 kb of rcDNA, which enters the nucleus. The positive strand of rcDNA is incomplete, so the genes encoded within it cannot be expressed; however, rcDNA is transformed to cccDNA in the nucleus. The transformation from rcDNA to cccDNA begins with the completion of DNA positive strand synthesis and continues with the removal of DNA polymerase and the primary RNA attached to the negative strand of DNA, leading to double-stranded linear DNA (dslDNA). A ligase enzyme binds both ends of dslDNA to form cccDNA. After that, cccDNA forms non-integrated cccDNA minichromosome by associating with host histones and HBc proteins [9,22,42–44].
 HBV consists of relaxed circular DNA (rcDNA), polymerase enzyme, capsid (HBcAg), non-structural antigen (HBeAg), and envelope (HBsAg). (b) HBV genetic material consists of four open reading frames (ORFs) encoding HBsAg, HBcAg, and HBeAg, polymerase, and HBx, and also three CpG islands. (c) The HBV life cycle begins when virions bind to sodium-taurocholate cotransporting polypeptide (NTCP). In the nucleus, rcDNA transforms into cccDNA minichromosome. The cccDNA minichromosome")
The structure, genetic material, life cycle, and formation of HBV cccDNA minichromosome. (a) HBV consists of relaxed circular DNA (rcDNA), polymerase enzyme, capsid (HBcAg), non-structural antigen (HBeAg), and envelope (HBsAg). (b) HBV genetic material consists of four open reading frames (ORFs) encoding HBsAg, HBcAg, and HBeAg, polymerase, and HBx, and also three CpG islands. (c) The HBV life cycle begins when virions bind to sodium-taurocholate cotransporting polypeptide (NTCP). In the nucleus, rcDNA transforms into cccDNA minichromosome. The cccDNA minichromosome's genes are transcribed and translated. After translation, HBx and HBc proteins re-enter the nucleus to regulate of cccDNA minichromosome and host genes through epigenetic mechanisms. These two proteins can affect the cccDNA minichromosome's conformation, stability, and transcription. The encapsidation process is also supported by HBc and polymerase. The genetic material in the form of pregenomic RNA (pgRNA) is then transformed into negative-strand DNA through the reverse transcription (RT) process. HBsAg is assembled into an envelope and covers the capsid. Mature HBV and HBeAg are secreted from the cell. Created with BioRender.com.
The HBV genome has six start codons, four promoters, and two transcription-enhancing elements (Enh) [8] with four main overlapping open reading frames (ORFs) (Fig. 2B). The first ORF consists of the PreS1, PreS2, and S genes, which encode for three different types of HBV surface antigen (HBsAg). The PreC and Core genes encode the core protein that forms the viral capsid (HBcAg) and the released e-antigen (HBeAg) in the second ORF. The third and fourth ORFs encode DNA polymerase and non-structural protein X (HBx), respectively [8,21]. In addition, the HBV genome has three CpG islands [8,9,45]. The CpG island I overlaps with the S gene. CpG island II has overlapping sequences with Enh I, Enh II, promoter X, and promoter C. CpG island III overlaps the PreS1 promoter and the polymerase start codon [8,9,46].
The cccDNA transcription process in the nucleus generates four types of mRNA with lengths of 3.5, 2.4, 2.1, and 0.7 kb [8], depending on transcription initiation from the different promoters [47,48]. The four mRNAs are translated as they leave the nucleus. The HBx protein returns to the nucleus to contribute to the epigenetic regulation of the cccDNA minichromosome, while HBeAg and HBsAg enter the endoplasmic reticulum and Golgi apparatus, respectively. HBeAg is secreted outside the cell, while HBsAg is assembled into the viral envelope. Some HBc proteins re-enter the nucleus, while others are used in the cytoplasm to form capsids [49,50]. Encapsidation occurs in the cytoplasm, involving HBcAg, DNA polymerase, and pre-genomic RNA (pgRNA). The reverse transcription (RT) process involves the reverse transcriptase enzyme to convert pgRNA into negative strand DNA, which is then used to generate a piece of positive-strand DNA. The nucleocapsid containing rcDNA is covered by a coat protein. New virus particles leave the cell by exocytosis [10,21,22].
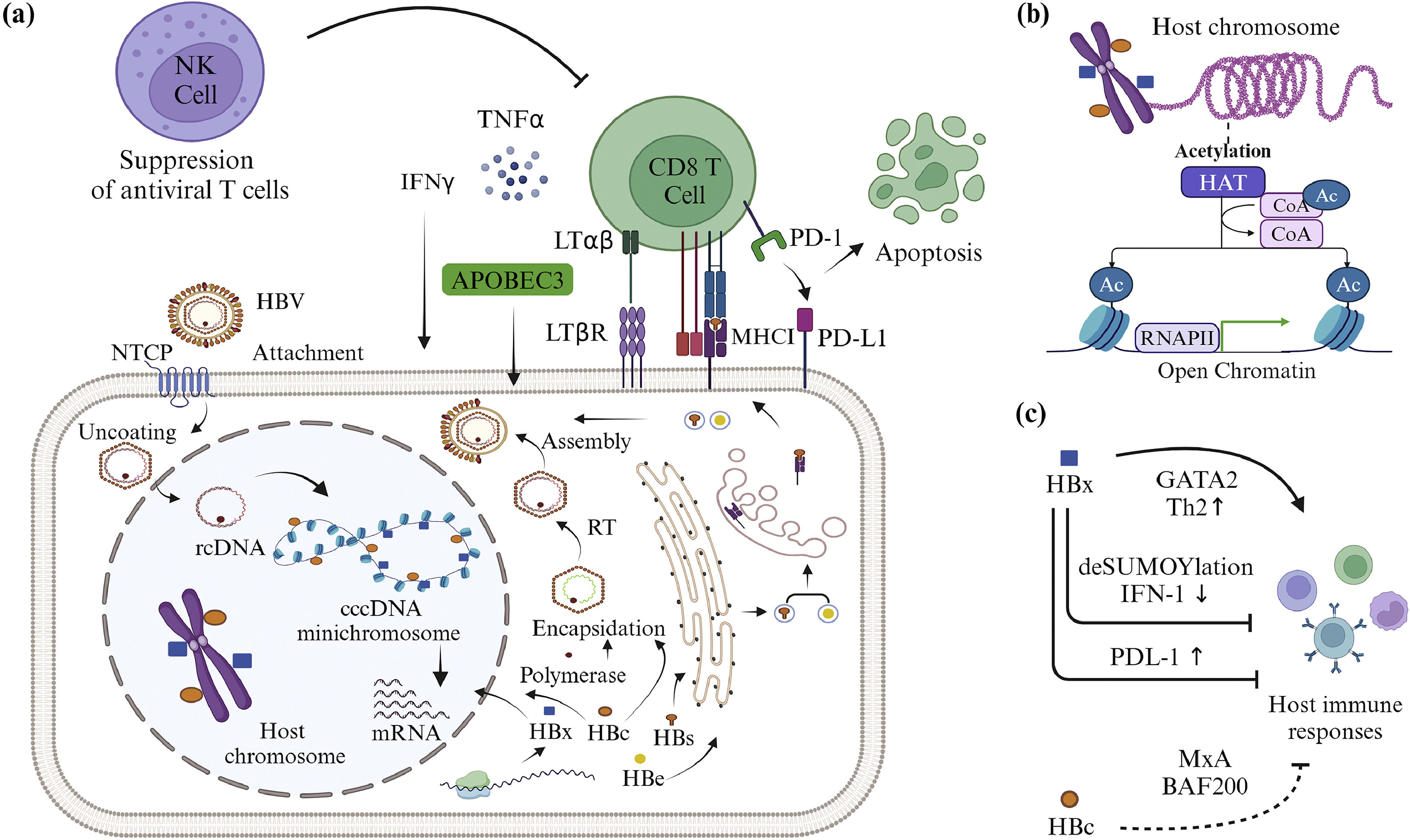
3Action of HBV and host immune systemImmature dendritic cells can recognize HBV when it enters the body. Dendritic cells will phagocytose and deliver HBV antigens to the cell surface on major histocompatibility complex (MHC) class I or II molecules. Mature dendritic cells will migrate to the lymph nodes and activate T cells. CD4 T cells (T Helper 2) will recognize an antigen on MHC class II. Th2 cells will activate B cells, leading them to differentiate into plasma cells by releasing interleukin 4 (IL-4) or IL-21 [51]. Plasma cells can produce antibodies against HBsAg (anti-HBs), anti-HBc, and anti-HBe [12,51,52]. The antibodies produced may combat HBV in several ways. Anti-HBs, for example, can inhibit HBV binding to NTCP while stimulating antibody-dependent cell-mediated cytotoxicity by natural killer (NK) cells and antibody-dependent cellular phagocytosis by Kupffer cells. B cells can produce both IL-6 and antibodies. These cytokines are known to suppress cccDNA transcription and acetylation, as well as NTCP expression [51,53,54].
If the antigen is present on MHC class I, CD8 T cells will recognize it and proliferate. CD8 T cells will recognize HBV antigens presented by infected liver cells on MHC class I (Fig. 3A). After specific recognition, CD8 T cells can eliminate HBV through two mechanisms: non-cytolytic and cytolytic [51,55]. In a non-cytolytic mechanism, CD8 T cells will release cytokines such as interferon γ (IFNγ) or tumor necrosis factor (TNF). Both cytokines can degrade cccDNA minichromosome by activating hepatocyte apolipoprotein B mRNA-editing enzyme 3 (APOBEC3) deaminase [51,56,57]. The interaction of lymphotoxin αβ (LTαβ) on CD8 T cells and lymphotoxin β receptor (LTβR) on liver cells can activate APOBEC3 [51,58]. APOBEC3 can cause hypermutation of cytosine bases to uracil (C→U) in HBV mRNA during the RT process [56,58]. This can interfere with HBV replication. CD8 T cells can eliminate HBV in liver cells without causing liver tissue damage through non-cytolytic mechanisms [51]. In contrast to the non-cytolytic process, the cytolytic mechanism of CD8 T cells leads to the formation of cytotoxic granules composed of granzyme B and perforin. Granzyme B can enter HBV-infected liver cells and remove cytochrome c from the mitochondria, as well as degrade liver cell DNA by activating procaspase [59–61]. The cytolytic process causes hepatocytes and the HBV inside them to lyse [51].
 When HBV replicates in cells, the liver can present the HBV antigen to the cell surface on major histocompatibility complex (MHC) class I. The CD8 T cell receptor recognizes the antigen specifically. CD8 T cells can secrete cytokines and activate apolipoprotein B mRNA-editing enzyme 3 (APOBEC3) to eliminate HBV without cytolysis, but this action can be inhibited by natural killer (NK) cells, which are abundant in the liver environment. T cells can potentially get exhausted as a result of excessive antigen exposure and high programmed death ligand-1 (PD-L1) expression, leading to a decrease in the number of T cells due to apoptosis. (b) HBc and HBx that re-enter the nucleus can alter the expression of host-immune-related genes via epigenetic mechanisms such as acetylation. (c) HBc inhibits IFN-induced host immune responses by binding to the myxovirus protein A (MxA) and Brahma‐related gene 1-associated factor 200 (BAF200) promoters. HBx can also inhibit interferon (IFN)-induced host immunological responses, produce T helper 2 (Th2) cells that do not participate in cellular immune responses, and promote T cell exhaustion by boosting PD-L1 expression. Created with BioRender.com.")
The interaction of HBV and the host immune system. (a) When HBV replicates in cells, the liver can present the HBV antigen to the cell surface on major histocompatibility complex (MHC) class I. The CD8 T cell receptor recognizes the antigen specifically. CD8 T cells can secrete cytokines and activate apolipoprotein B mRNA-editing enzyme 3 (APOBEC3) to eliminate HBV without cytolysis, but this action can be inhibited by natural killer (NK) cells, which are abundant in the liver environment. T cells can potentially get exhausted as a result of excessive antigen exposure and high programmed death ligand-1 (PD-L1) expression, leading to a decrease in the number of T cells due to apoptosis. (b) HBc and HBx that re-enter the nucleus can alter the expression of host-immune-related genes via epigenetic mechanisms such as acetylation. (c) HBc inhibits IFN-induced host immune responses by binding to the myxovirus protein A (MxA) and Brahma‐related gene 1-associated factor 200 (BAF200) promoters. HBx can also inhibit interferon (IFN)-induced host immunological responses, produce T helper 2 (Th2) cells that do not participate in cellular immune responses, and promote T cell exhaustion by boosting PD-L1 expression. Created with BioRender.com.
Although the immune system has mechanisms to combat HBV, the virus can evade the immune response. Two HBV antigens, the HBc and HBx proteins, affect the epigenetics of host cells, suppressing the expression of host immunity-related genes (Figs. 3B and C). HBc can decrease the expression of the myxovirus protein A (MxA) gene [9,62]. MxA inhibits HBV transcription by interfering with polymerase activity. IFN stimulation can activate transcription of the MxA gene. IFN can phosphorylate the signal transducer and activator of transcription 1 (STAT1) transcription factor, increasing its ability to bind to the MxA promoter. When HBc binds to the MxA gene promoter, it acetylates the STAT1 transcription factor, causing the STAT1 transcription factor to dephosphorylate and lose its ability to bind to the promoter [49,63]. HBc can also bind to Brahma‐related gene 1-associated factor 200 (BAF200), inhibiting the synthesis of interferon-induced transmembrane protein 1, and preventing the virus entry into the cytoplasm [9,64].
HBx is demonstrated to remove transcription factors from nuclear domain 10 by a deSUMOylation mechanism. This mechanism involves proteases, leading to the inactivation of SUMO [65]. This deSUMOylation inhibits IFN-I response pathway components and modifies host epigenetics through transcription coactivator 300 or histone deacetylase 1 (HDAC1) [9,66]. HBx can also influence the direction of Th cell induction. HBx can increase the expression of ST2 receptors, causing the induction of Th2 cells in the presence of IL-33 [9,67]. HBx activates ST2 expression through histone acetyltransferase (HAT), by acetylating the transcription factor GATA-binding factor 2 (GATA2). GATA2 acetylation can enhance its ability to bind to DNA [68].
HBx has been suggested as one of the causes of T and B cell exhaustion and dysfunction. Several studies have shown that when CHB patients were exposed to high levels of HBV antigens over a long period of time, exhaustion and dysfunction in T and B cells were noticed [24,15,69]. Exhaustion can reduce T cell proliferation and cytotoxicity [12,51,70], as well as decrease B cell differentiation and defective antibody production [24]. Exhausted T and B cells can produce an overabundance of immunosuppressive receptors, including programmed death protein-1 (PD-1) [15,71]. HBx can cause hepatocytes to express more programmed death ligand-1 (PD-L1), resulting in higher PD-L1 binding to the PD-1 receptor (Fig. 3A). HBx increases PD-L1 expression through the phosphatase and tensin homolog deleted on chromosome 10 (PTEN)/β-catenin/c-Myc signaling pathway [72]. HBx can reduce PTEN expression, leading to increased β-catenin/c-Myc signaling. Epigenetic factors such as hypermethylation of the PTEN promoter or deacetylation of histones have a significant impact on decreased PTEN expression. Binding of PD-1 and PD-L1 has been shown to increase the activity of the transcription factors thymocyte selection-associated high mobility group box [12,73,74] and Bcl2-interacting mediator [12,51,75,76] and also cause mitochondrial metabolism disruptions [12,51,77]. Furthermore, T cell exhaustion can cause the production of TNF and interleukin 12 (IL-12), which are T cell activators, NK, and NKT cell stimulators [69]. NK cells are known to cause apoptosis in T cells by expressing the TNF-related apoptosis-inducing ligand [51,78].
4Epigenetic regulation in HBV cccDNA and hosts leading to HCCEpigenetic regulatory mechanisms alter the stability, conformation, and transcription of cccDNA minichromosome. Several host cell proteins and two HBV proteins, HBc and HBx, contribute to the epigenetic regulation of HBV cccDNA [8–11,21,22]. These two HBV proteins have been reported to interact with host enzymes involved in DNA methylation, including DNA methyltransferase (DNMT), several histone-modifying enzymes such as HAT and HDAC [9,49], and serine arginine protein kinase 1 [79]. The HBc protein, a 21-kDa structural polypeptide, is a non-histone protein that can bind directly to cccDNA's C-terminal domain [9,10,49,80]. It controls the arrangement of nucleosomes on the cccDNA minichromosome, allowing it to have a more compact structure than host cell chromatin [8,49,81].
HBc may stimulate cccDNA transcription through hypomethylation in the CpG island II region [9,10,49,82]. HBc may also recruit HATs to bind to histones on the cccDNA minichromosome, such as CREB-binding protein, p300, and a factor related to both. This stimulates histone acetylation and modifies the conformation of the chromosomes, which were initially very compact, to become looser. This condition allows the transcription of genes encoded in the cccDNA minichromosome [9,10,83]. Besides, HBc may increase the binding of NF-KB to EnhII by acetylation and methylation. Pre-C gene transcription can be increased by NF-KB binding to EnhII [9,49,84].
HBx is a small oncogenic protein with a molecular weight of 17 kDa [9,85] that has multiple roles in cccDNA transcription. HBx, like HBc, may open a minichromosome configuration by recruiting HAT [10] and causing hypomethylation at different sites, especially on CpG islands II and III [9,11,86–88]. The opening of the minichromosome configuration allows Parvulin 14 (Par14) and Par17, which are not phosphorylated in the serine region, to bind to cccDNA. Negatively charged residues in Par14 and Par17 can bind to HBx, increasing its affinity for cccDNA. The binding of Par14 and Par17 to the Enh region of cccDNA has been shown to increase cccDNA transcription [89]. HBx has been shown to reduce hypoacetylation of histones that bind to cccDNA and to regulate HDAC1 attachment to cccDNA. Hypoacetylation of histones H3 and H4 and the HDAC1 binding to cccDNA may inhibit cccDNA transcription [11].
Furthermore, HBx may inhibit sirtuin 3 from binding to cccDNA. Sirtuin 3 has been found to interact with histone methyltransferases, such as the SET domain-containing protein 1A, to eliminate H3K9ac and increase H3K9me3 [90]. HBx has also been shown to inhibit the methyltransferase activity of the arginine N-methyltransferase. This enzyme suppresses cccDNA transcription by methylating transcription factors [91]. HBx is considered to be able to maintain the structure of the cccDNA minichromosome. HBx can bind to damaged DNA-binding protein 1 and inhibit SMC5/6. The SMC5/6 complex will be degraded because of its interaction with the ubiquitin proteasome system. The SMC5/6 group is a host restriction factor that inhibits cccDNA transcription [8,9,11,92].
Aside from their role in the epigenetic regulation of cccDNA, HBc and HBx can also influence the epigenetics of the host cells. These two proteins can inhibit the expression of tumor suppressor genes in host cells by DNA methylation and histone modification, contributing to the development of HCC [11]. HBc may contribute to the development of HCC by suppressing apoptosis in liver cells. HBc interacts with the transcription factor E2F1 [9,49,93], causing phosphorylation and the loss of E2F1′s ability to bind to the p53 gene promoter [94]. The p53 gene encodes the p53 protein, which can cause cell cycle arrest and apoptosis in response to DNA damage, hypoxia, and oncogene activation [95]. Apart from p53, HBc has been shown to bind to the death receptor 5 promoter, suppressing transcription and decreasing cell apoptosis by the TNF-related apoptosis-inducing ligand pathway. Another study demonstrated that HBc facilitates the binding of APOBEC3A to cccDNA, which causes deamination and contributes to the development of HCC [56,96].
HBx has been shown to bind to the promoters of several host genes, including cancer-related genes. HBx, in conjuction with DNMT activity, has the potential to induce hypermethylation or hypomethylation of several tumor suppressor genes [97]. Hypomethylation and the binding of HBx and HDAC1 to the promoter may prevent the synthesis of tumor-promoting regulators [86]. For example, HBx has been shown to effectively bind DNMT1 and DNMT3A to the promoter and decrease the expression of the mediator p53 [11]. HBx can also decrease p53 binding to the metastasis-associated protein 1 promoter, increasing metastasis-associated protein 1 synthesis and enhancing HCC metastasis [86]. Several studies have found that HBx can reduce the expression of tumor suppressor genes such as suppressors of cytokine signaling-1 [98], Ras-association domain family 1A [99], procadherin-10 [100] and E-cadherin [101] by modifying DNA methylation.
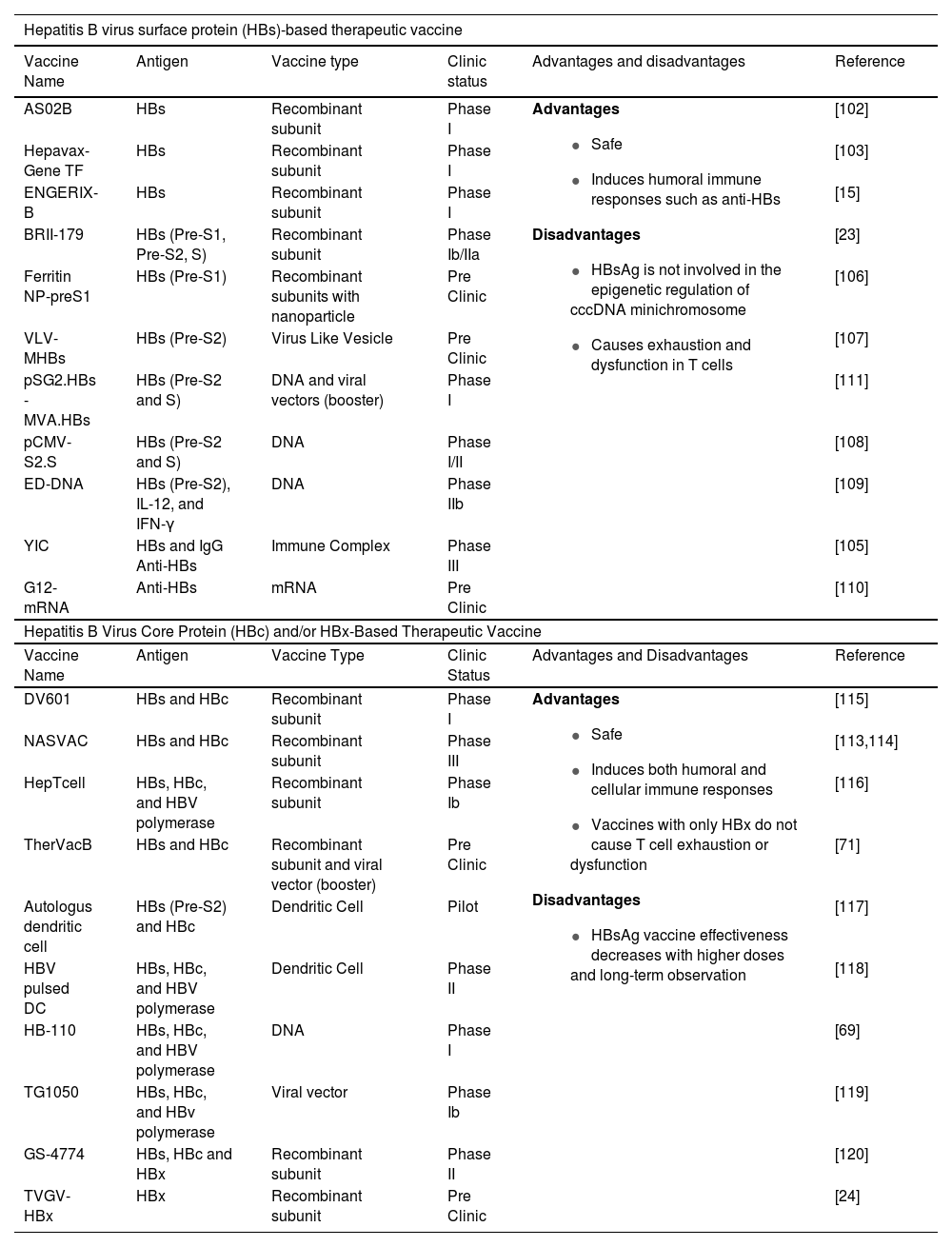
5Therapeutic vaccine for CHBAt the beginning of its development in 1994, vaccine therapy used prophylactic vaccine containing HBsAg. It was administered to CHB patients and was effective in decreasing HBV DNA and leading to the development of anti-HBe in some patients [13,38]. Therefore, various therapeutic vaccines containing HBsAg are under development (Table 1). Although HBsAg is used as in a prophylactic vaccine, different adjuvants was prepared in therapeutic vaccines [14]. HBsAg-based vaccines are used to treat CHB at doses ranging from 10 micrograms to 40 micrograms. The frequency of vaccination injections was three to twelve times higher than prophylactis vaccination [38]. The theurapetic vaccine can be also injected at various sites, in particular intradermally [14].
Therapeutic vaccine development for chronic hepatitis B in the Last Two Decades.
| Hepatitis B virus surface protein (HBs)-based therapeutic vaccine | |||||
|---|---|---|---|---|---|
| Vaccine Name | Antigen | Vaccine type | Clinic status | Advantages and disadvantages | Reference |
| AS02B | HBs | Recombinant subunit | Phase I | Advantages
| [102] |
| Hepavax-Gene TF | HBs | Recombinant subunit | Phase I | [103] | |
| ENGERIX-B | HBs | Recombinant subunit | Phase I | [15] | |
| BRII-179 | HBs (Pre-S1, Pre-S2, S) | Recombinant subunit | Phase Ib/IIa | [23] | |
| Ferritin NP-preS1 | HBs (Pre-S1) | Recombinant subunits with nanoparticle | Pre Clinic | [106] | |
| VLV-MHBs | HBs (Pre-S2) | Virus Like Vesicle | Pre Clinic | [107] | |
| pSG2.HBs - MVA.HBs | HBs (Pre-S2 and S) | DNA and viral vectors (booster) | Phase I | [111] | |
| pCMV-S2.S | HBs (Pre-S2 and S) | DNA | Phase I/II | [108] | |
| ED-DNA | HBs (Pre-S2), IL-12, and IFN-γ | DNA | Phase IIb | [109] | |
| YIC | HBs and IgG Anti-HBs | Immune Complex | Phase III | [105] | |
| G12-mRNA | Anti-HBs | mRNA | Pre Clinic | [110] | |
| Hepatitis B Virus Core Protein (HBc) and/or HBx-Based Therapeutic Vaccine | |||||
| Vaccine Name | Antigen | Vaccine Type | Clinic Status | Advantages and Disadvantages | Reference |
| DV601 | HBs and HBc | Recombinant subunit | Phase I | Advantages
| [115] |
| NASVAC | HBs and HBc | Recombinant subunit | Phase III | [113,114] | |
| HepTcell | HBs, HBc, and HBV polymerase | Recombinant subunit | Phase Ib | [116] | |
| TherVacB | HBs and HBc | Recombinant subunit and viral vector (booster) | Pre Clinic | [71] | |
| Autologus dendritic cell | HBs (Pre-S2) and HBc | Dendritic Cell | Pilot | [117] | |
| HBV pulsed DC | HBs, HBc, and HBV polymerase | Dendritic Cell | Phase II | [118] | |
| HB-110 | HBs, HBc, and HBV polymerase | DNA | Phase I | [69] | |
| TG1050 | HBs, HBc, and HBv polymerase | Viral vector | Phase Ib | [119] | |
| GS-4774 | HBs, HBc and HBx | Recombinant subunit | Phase II | [120] | |
| TVGV-HBx | HBx | Recombinant subunit | Pre Clinic | [24] | |
Several studies have developed recombinant HBsAg-based therapeutic vaccines during the last 16 years, including AS02B, Hepavax-Gene TF, ENGERIX-B, BRII-179, and Ferritin NP-PreS1 (Table 1). However, patients treated with the AS02B vaccine had no different effects than those who received Lamivudine only [102]. Patients with low HBsAg levels who received multiple doses of the ENGERIX-B vaccination demonstrated a significant HBsAg level reduction at 48 weeks post-treatment; however, detection of HBV DNA was not performed [15]. In contrast, patients who received NA treatment plus the Hepavax-Gene TF vaccine demonstrated a reduction in HBV DNA but not in HBsAg [103]. The BRII-179 vaccine stimulates antibody and T cell responses without affecting the patient's virological status [23]. A Chinese research team attempted to combine the recombinant HBsAg complex from yeast cells with high-titer anti-HBs IgG (rHBsAg-IgG) to increase the efficiency of the recombinant HBsAg vaccine [104,105]. Based on clinical trials, YIC has successfully induced cytolytic and non-cytolytic responses, resulting in decreased levels of HBsAg and HBV DNA. In phase III clinical studies, however, the vaccine's efficacy declined as the vaccine dose increased [105].
Several studies have also attempted to improve the efficiency of the recombinant HBsAg vaccination using delivery mechanisms such as nanoparticles and virus-like vesicles. The Ferritin NP-preS1 vaccine uses ferritin nanoparticles to precisely deliver the Pre-S1 protein to dendritic cells [106], while the VLV-MHBs use the Vesicular Stomatitis Virus [107]. Both vaccinations had more potential outcomes where they could induce an antibody response and reduce the amount of HBsAg, HBV DNA, and RNA. However, there are no data available in clinical trials.
Aside from the recombinant subunit vaccination, various studies are working on the HBsAg DNA vaccine. The pCMV-S2.S vaccine could not prevent recurrence, and only a few patients had reduced HBV DNA [108]. The ED-DNA vaccination decreased HBsAg levels at the beginning of therapy, but the levels increased again after a long period of observation [109]. Although ongoing research develops the HBsAg DNA vaccine using the Modified Vaccinia Virus Ankara, which encodes HBsAg, it is still unable to control HBV infection [109]. The latest study also developed a G12-mRNA vaccine encoding anti-HBs. This vaccination induces long-term HBsAg seroclearance and has the potential to contribute to immune system support, but the only parameter for therapeutic success was HBsAg, and the observation period was 30 days [110].
Based on the data presented, it seems that vaccines containing HBsAg as the primary constituent has limited efficacy for CHB treatment. The amount of HBsAg and HBV DNA in patients rises with increasing doses or with long-term observation [103,105,108,109]. This indicates that HBV cccDNA is still present in the cell and the cccDNA transcription process is still active. HBsAg is not involved in the epigenetic control of cccDNA minichromosome. Furthermore, HBV produces a high amount of HBsAg, which is released into the blood [24]. Before receiving vaccination therapy, the patient had been exposed to this antigen in significant amounts and over a long period; thus T cells will be exhausted and dysfunctional as a result of the HBsAg-based vaccination treatment.
HBsAg can stimulate the production of neutralizing antibodies [111]. This immune response is suitable for infection prevention (prophylactic vaccination) but insufficient for treatment, which requires the involvement of T cells, specifically CD8 T cells, in eliminating infected liver cells [112]. Several studies have found that DNA vaccines may induce a functional immune response profile similar to the natural immune response in HBV-infected patients. DNA vaccines can stimulate the production of antibodies, T Helper 1 cells, T cells that produce IFN γ, and NK cells that produce various cytokines. The HBsAg DNA vaccine, however, remains ineffective at inducing a cellular immune response capable of eliminating cccDNA.
Due to the limitations of HBsAg-based therapeutic vaccines, researchers began combining HBsAg with other antigens involved in epigenetic regulation of the cccDNA minichromosome to improve vaccine efficacy. Several studies have used HBsAg and HBcAg as the main components of therapeutic vaccines. The DV601 and NASVAC vaccines contain recombinant HBsAg and HBcAg. Although both vaccines were shown to activate adaptive and innate immune responses, produce anti-HBs and anti-HBe, and reduce the amount of HBsAg, HBeAg, and HBV DNA in patients, only NASVAC continued phase III clinical trials [113–115]. However, NASVAC was unable to completely eliminate HBV DNA; therefore, the amount of HBV DNA in patients increased after 3 years [113]. Other experiments involving HBV polymerase in the therapeutic vaccine composition showed no better results, and even HepTcell was unable to decrease the HBsAg levels [116]. Several studies have attempted to develop vaccines containing dendritic cells that express HBsAg and HBcAg to boost the immune response of CHB patients. HBV DNA decreased in patients with autologous dendritic cells and HBV pulsed DC vaccines, but there was no significant reduction in the level of both HBsAg and HBeAg [117,118].
Regarding DNA vaccines, the HB-100 and HB-110 vaccines contain DNA encoding HBs, HBc, and HBV polymerase. In HB-110, codon optimization and deletion of the HBx oncogene were performed to improve the vaccine's immunogenicity and efficacy. However, the limitation of the study was that vaccines were tested on different groups of people from various ethnic backgrounds. HB-100 was examined on Caucasians in phase I clinical trials, while HB-110 was tested on Asians, hence their efficacy cannot be compared. Results showed that HB-100 is more effective than HB-110. On the other hand, HB-110 was successful in generating higher titers of T cells and antibodies in mice at the pre-clinical stage. This discrepancy might be caused due to difference among route of HBV transmission between Caucasians and Asians. The majority of Asians transmit HBV vertically, exposing their bodies to HBV antigens from a young ages [69].
In addition, the TG1050 vaccine was developed based on adenovirus serotype 5 expressing HBs, HBc, and HBV polymerase. The results of the study showed a decrease in serum levels of HBcAg, but not a significant reduction in the level of HBsAg [119]. According to current studies, using a booster like Modified Vaccinia Virus Ankara that can express HBsAg, HBcAg, and siRNA enhances the efficiency of the TherVacB vaccine. siRNA inhibits the activity of PD-1 through epigenetic mechanisms. Pre-clinical studies have shown that TherVacB boosts T cell responses, effectively reduces antigens in the blood, regulates HBV replication, eliminates infected hepatocytes from the liver, and significantly decreases HBV pgRNA [71].
Although few studies used HBx as the main component in therapeutic vaccines, at least two trials have been successful in combining HBx with other antigens. The GS-4774 vaccine, a therapeutic vaccine for CHB, contains recombinant HBsAg, HBcAg, and HBx [120]. In phase II clinical trials, GS-4774 increased the number of T cells while decreasing the amount of HBsAg in the blood, although no patients reported a decrease in HBsAg levels by week 48. Unlike prior vaccines, TVGV-HBx is a therapeutic vaccine that only contains recombinant HBx as its main component [24]. The vaccine effectively reduced HBV DNA, induced CD8 and CD4 T cells, and eliminated HBsAg in mice. Without a booster, vaccine protection in test animals lasted for 30 days.
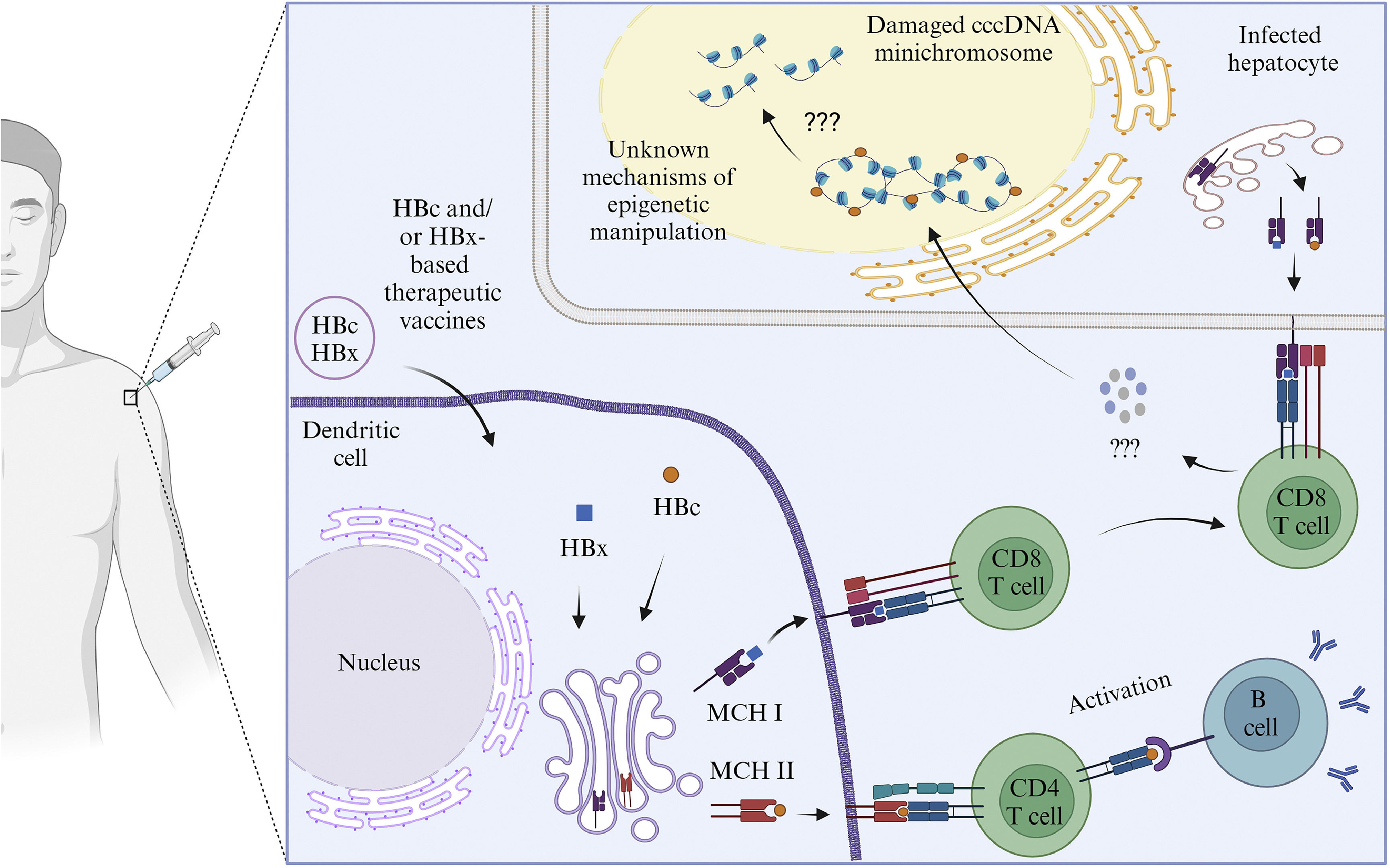
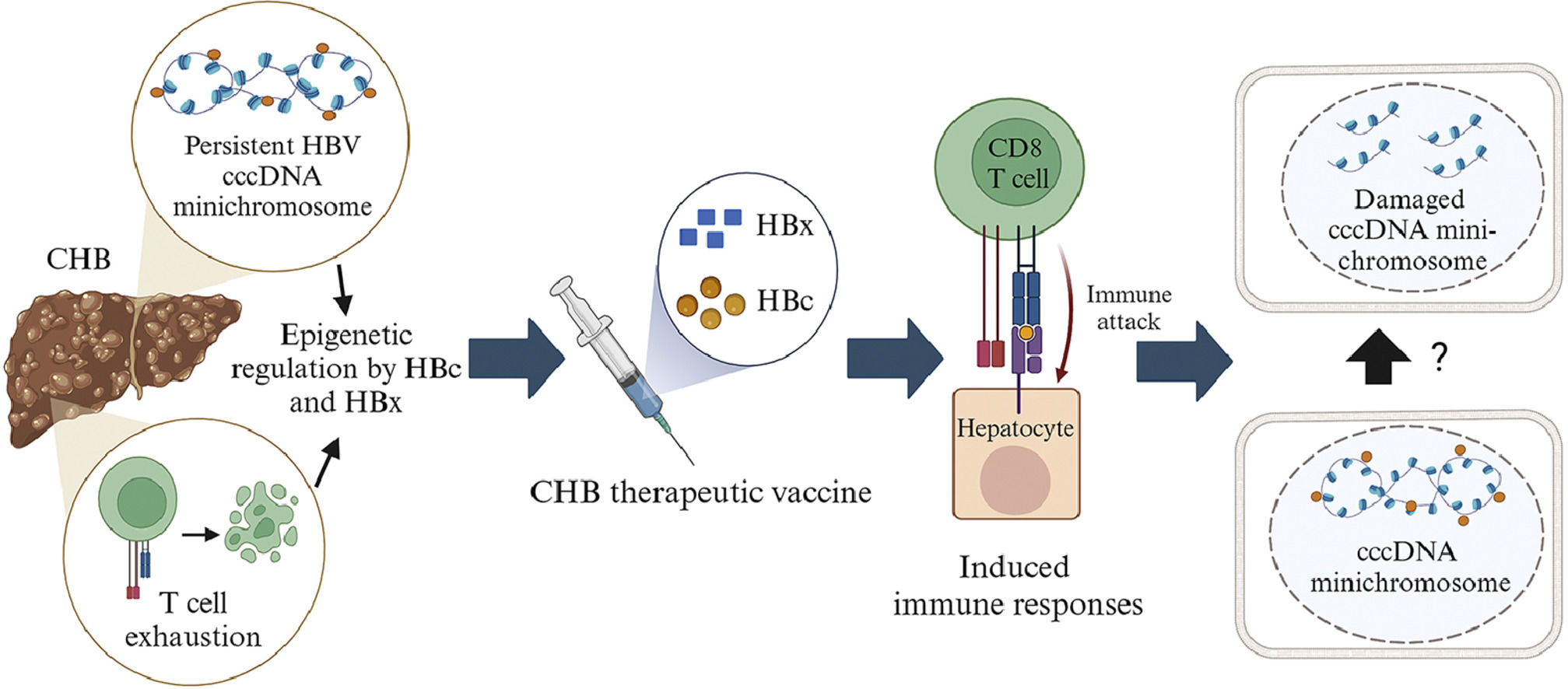
All HBc and/or HBx-based therapeutic vaccines are safe, and the majority can reduce HBV DNA and HBsAg levels. These therapeutic vaccines have a higher potential of effectiveness than those containing only HBs [24,71,113–116,119,120]. The TVGV-HBx vaccine with a CpG oligodeoxynucleotide adjuvant can induce innate and adaptive immunological responses in CD8 and CD4 T cells without causing exhaustion and dysfunction[24]. Several studies have found that two HBV proteins, HBc and HBx, are involved in the epigenetic regulation of cccDNA minichromosome. Furthermore, both proteins can alter the epigenetics of host cells, inhibiting the expression of host genes related to immunity and tumor suppressors [8–11]. These two proteins are also considered to have a significant role in the development of HCC. Unfortunately, there is no indication that the immune response induced by HBc and/or HBx-based therapeutic vaccines may modify the epigenetics of the cccDNA minichromosome (Fig. 4). Furthermore, therapeutic vaccines based on HBc and/or HBx still have limitations. HBsAg is still present in several therapeutic vaccinations based on HBc and/or HBx. The presence of HBsAg in therapeutic vaccines causes T cell exhaustion and dysfunction, leading to a decrease in vaccination efficacy when the dose increases or long-term monitoring is performed [69,71,113,114]. This limitation is one of the reasons why few HBc and/or HBx-based therapeutic vaccines have progressed to phase III clinical trials.

The illustration of the mechanism of HBc and/or HBx-based mRNA therapeutic vaccines. HBc and/or HBx-based vaccines will enter the dendritic cells. HBc and/or HBx antigens will be presented to the cell surface with the help of MHC class I and class II proteins. Antigens presented by MHC class II will be recognized by CD4 T cell receptors, activating B cells to produce antibodies. Antigens presented by MHC class I will be recognized by CD8 T cell receptors which will recognize antigens presented by infected liver cells. The CD8 T cells may secrete cytokines that attack HBc and/or HBx. Damage to HBc and/or HBX may lead to conformational, stability and translational disruption of the minichromosome cccDNA through unknown epigenetic mechanisms. Created with BioRender.com.
Hundreds of million people are still living with chronic HBV infection worldwide. A significant number of new infections each year is still observed. Regardless major prevention efforts via nationwide vaccination and the administration of antiviral therapies to HBV-infected people, CHB led to an estimated 800,000 deaths, mostly from liver cirrhosis and HCC [121,122].
The severity and the clinical onset of the disease is related to both viral and host factors. The persistency of HBV cccDNA in the nucleus of infected cells is a critical step in the CHB infection. While current treatments for CHB infection are powerful in suppressing viral replication, they are unable to remove and or eliminate cccDNA effectively. When antiviral treatment is interrupted, and in immunosuppressed conditions related to host immunologic problems, the persistence of cccDNA might induce HBV reactivation. Therefore, eliminating cccDNA and its underlying mechanisms is one of the key concepts to inhibit CHB progression and to achieve a true cure.
In addition, CHB is well-known immunological disease. Apart of the direct oncogenic mechanism of the HBV itself, such as HBV DNA integration into the host genome, HBV pathogenesis in triggering liver damage is a result of an indirect mechanism related to necro-inflammation and immunological response of the host (reviewed in [123]). Thus, in order to look for a potential for CHB cure, a strategy on the combination of modulation of both viral (cccDNA) and host (immunological response) is a logical approach. Hence, therapeutic vaccine can be a valid treatment against CHB, and in longer perspective, against the more severe HBV-related liver diseases.
In this review, we showed vast evidences that HBc and HBx are two HBV proteins involved in the epigenetic regulation of cccDNA minichromosome. Furthermore, both proteins can alter the epigenetics of host cells, inhibiting the expression of host genes related to immunity and tumor suppressors [8–11]. Regardless the limited data available of the effect of these vaccines on the persistency of cccDNA, and only few candidates have been assessed in clinical trials, advances and technologies in vaccine development and growing number of vaccine platforms will be give an important contribution in the field.
Vast developments in drug discovery and molecular biology techniques gives an important breakthrough in drug development, both as a single agent or in combination therapies. Several studies [124,125] focusing on innovative strategies targeting the cccDNA had been conducted, involving either disrupting cccDNA expression through gene editing techniques or employing epigenetic modifications to silence its transcription [124–126]. The potential of synthetic RNAi that had been demonstrated in both HBe-negative and -positive CHB patients [127]. In addition, by silencing the pgRNAs through RNAi trigger, viral replication can be suppressed and the formation of cccDNA can be indirectly hindered [127]. Additionally, in vitro study using CRISPR/Cas9 system inhibited both HBV antigen expression and replication, excised the entire full-length of integrated HBV genome, and disrupted cccDNA [128]. These molecular-targeted agent, combined together with neutralizing therapeutic vaccines, for example those against HBc and/or HBx, can be one of the promising options to target CHB, both for the virus itself and for the host immunological response.
To summarize, despite the challenges, the development of therapeutic vaccines offers new hope for CHB patients. The selection of HBV antigens is critical in the development of therapeutic vaccines for CHB. Antigens used as primary components in therapeutic vaccines must induce immune responses targeting proteins involved in the epigenetic regulation of HBV cccDNA without causing T cell exhaustion. The therapeutic vaccines, as single agents or in combination with other therapeutical regimens, are potential. Thus, multisectoral collaboration among scientists, clinicians, industries, and regulatory bodies is crucial in the HBV vaccine development in the future.
Author contributionsConceptualization: EAG, MIT, PGN, CS. Methodology: PGN, MIT, EAG. Investigation: PGN, MIT, AA, CS, EAG. Resources: AA, EAG. Writing-original draft preparation: PGN, AA, CS. Writing-review and editing: PGN, MIT, AA, CS, EAG. All authors have read and agreed to the published version of the manuscript.
FundingThis work was suported by Riset Unggulan of Institut Teknologi Bandung [grant numbers P-SITH.PN-6-06-2024] and Rumah Program 2024 of the Research Organization for Health, BRIN of Indonesia.
CS received a 2024 grant from the Fondazione Veronesi, Milan, Italy. We would also like to thank Aluicia Anita Artarini and Husna Nugrahapraja for the discussion about epigenetic manipulation-based hepatitis B vaccines. All concept figures were created with the Bio-Render illustration tool.



