



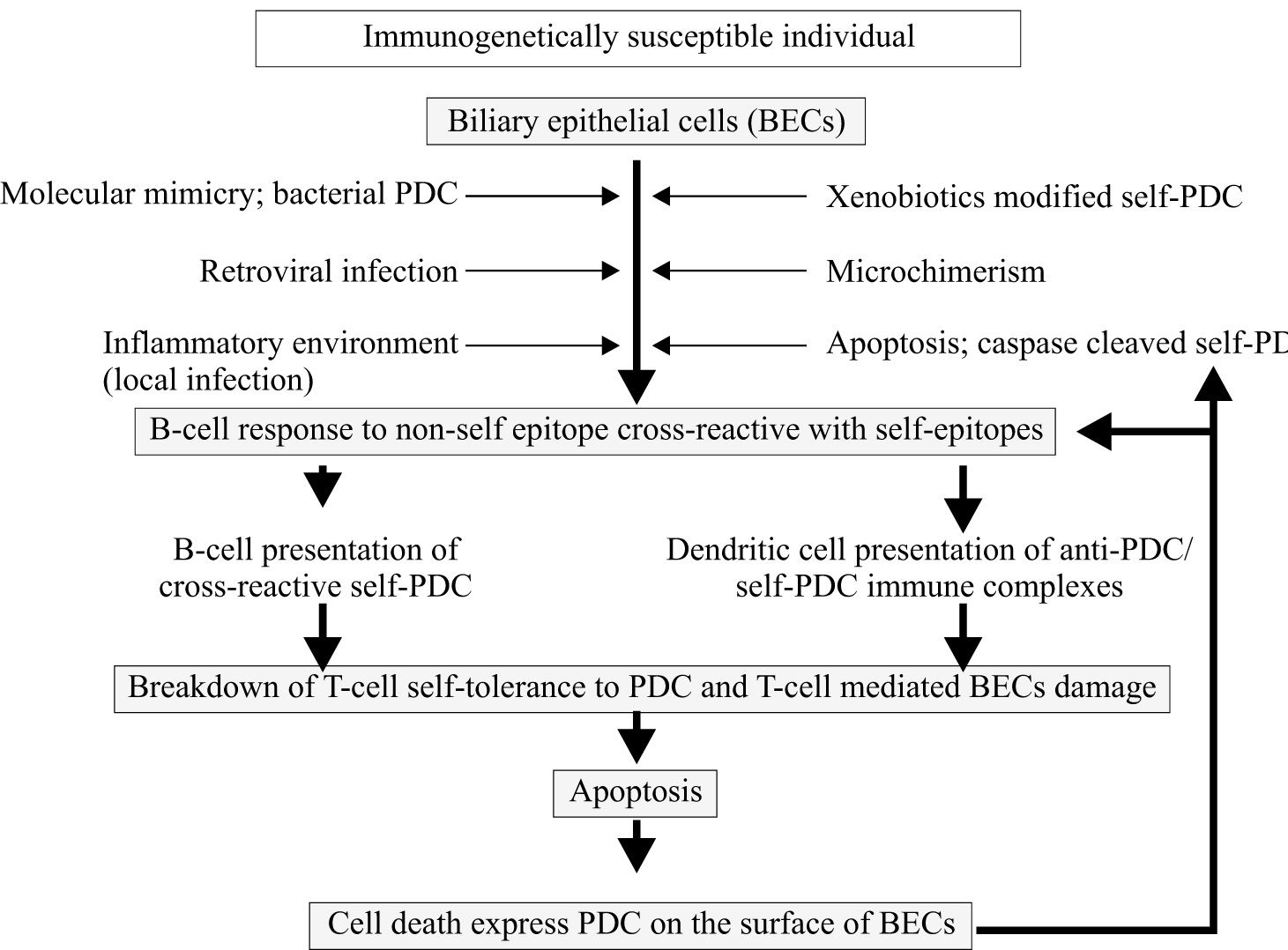

Primary biliary cirrhosis (PBC) is an organ-specific autoimmune disease that predominantly affects women and is characterized by chronic, progressive destruction of small intrahepatic bile duct with portal inflammation and ultimately fibrosis, leading to liver failure in the absence of treatment. The serologic hallmark of PBC is the presence of autoantibodies to mitochondria, especially to the E2 component of the pyruvate dehydrogenase complex (PDC). Current theories on the pathogenesis of PBC favor the hypothesis that the disease develops as a result of an inappropriate immune response following stimulation by an environmental or infectious agent. Like other better characterized autoimmune diseases, there appears to be a genetic susceptibility and a triggering event that initiates the autoimmune attack on bile duct cells. DRB1*0801 and DRB1*0803 are the major susceptibility alleles among Northern European and Japanese populations, respectively. The generation of immune responsiveness to self-antigen can result in pathogenic autoimmune damage of the intrahepatic biliary epithelial cells mediated by both humoral and cellular immune responses. The pathogenetic mechanism is believed to be caused by a defect in immunologic tolerance, resulting in the activation and expansion of self-antigen specific T and B lymphocyte clones and the production of circulating autoantibodies in addition to a myriad of cytokines and other inflammatory mediators. Human and animal studies have suggested that the induction of an antibody response reactive with self-antigen may result from a number of different priming events. Among the events demonstrated to induce an antibody response cross-reactive with self-PDC are exposures to bacterial PDC or retroviral proteins or xenobiotics or microchimerism. The diversity of the potential events giving rise to antibody responses cross-reactive with PDC, which could promote subsequent T-cell tolerance breakdown, suggests the intriguing possibility that PBC could represent a condition with a common final pathway but with multiple triggers able to induce a B-cell response cross-reactive with self-PDC. There are important questions about the pathogenesis of PBC which remain unanswered.
Primary biliary cirrhosis (PBC) is a chronic, progressive cholestatic liver disease of unknown cause that usually affects middle-aged women who are commonly diagnosed after evaluation for symptomatic elevations in serum hepatic biochemical parameters. Fatigue, pruritus, or unexplained hyperlipidemia may also be found at initial presentation. PBC affects all races and has no specific geographic predilection. Recent data from Olmsted County, Minnesota,1 suggest a stable incidence rate over the last 25 years but a higher prevalence than described in Canada.2 Overall incidence and prevalence rates of 27 and 402 cases per million population,1 respectively, are comparable to results from northern England.2 PBC progresses in most cases, but the rate of progression varies greatly among individual patients. Asymptomatic patients have longer life expectancies than symptomatic ones, but their survival is still less than that of healthy patients matched for age and sex if untreated.4,5 Anti-mitochondrial antibodies are present in 95 percent of patients long before clinical signs or symptoms appear. The disease is characterized histologically by the destruction of small intrahepatic bile ducts, portal inflammation, and progressive scarring. The diagnosis should be considered in the setting of elevated alkaline phosphatase levels, hypercholesterolemia, and IgM elevation. The presence of an antimito-chondrial antibody is highly characteristic of this disease. Recognition of the disease is important because effective medical therapy with ursodeoxycholic acid has been demonstrated to halt disease progression and improve survival independent of liver transplantation.6
The pathogenesis of primary biliary cirrhosisCurrent concepts in the pathogenesis of PBC favor the hypothesis that the disease develops as a result of an inappropriate immune response (in genetically predisposed individuals) following stimulation by an environmental factor (microorganisms or xenobiotics) as shown in Figure 1. Evidence suggests that PBC has a significant autoimmune component to its pathogenesis. Highly specific autoantibodies reactive with biliary epithelial cell surface antigens and associated autoimmune disorders observed among patients with PBC have also been cited as further evidence for an immunologic cause. PBC is a typical example of an organ specific autoimmune disease. Tissue damage is largely restricted to the intrahepatic biliary epithelial cells (BECs), associated extrahepatic abnormalities being essentially limited to the salivary and lacrimal glands (secondary Sjögrens syndrome), thyroid gland (Hashimoto’s thyroiditis), and skin (scleroderma). The inflammatory response is believed to result from the aberrant recognition of mitochondrial self-antigen by the immune system. A major advance towards defining the pathogenesis of PBC has been the precise identification of a mitochondrial protein that is the target of such autoantibodies and T lymphocyte responses. The breakdown of immunologic tolerance results in the activation and expansion of self-antigen specific T and B lymphocyte clones and the production of circulating autoantibodies in addition to a myriad of cytokines and other inflammatory mediators. Furthermore, apoptotic BEC have been found in PBC liver which is assumed to be specific for the target tissue.

Current theories on the pathogenesis of PBC favor the hypothesis that the disease develops as a result of an inappropriate immune response following stimulation by an environmental or infectious agent. The pathogenetic mechanism is believed to be caused by a defect in immunologic tolerance, resulting in the activation and expansion of self-antigen specific T and B lymphocyte clones and the production of circulating autoantibodies in addition to a myriad of cytokines and other inflammatory mediators. The induction of an antibody response reactive with self-antigen may result from a number of different priming events including exposure to bacterial PDC or retroviral proteins or xenobiotics or microchimerism or apoptotic cell express PDC that induce an antibody response cross-reactive with self-PDC.
A puzzling feature of PBC is similar to certain other autoimmune diseases. First, what is the precise cause of PBC? The presence of a detectable AMA might be an epiphenomenon that appears after some other triggering event. Secondly, why is the autoimmune attack predominantly organ-specific but the disease seemingly characterized by autoreactive immune responses directed at ubiquitous self-antigens. Thirdly, how does such breakdown in T-cell tolerance to self-antigen occur in PBC patients? These have led some to doubt whether autoreactive immune responses in fact play any meaningful role in the pathogenesis of PBC. The aim of this review is to summarize the current state of understanding of the pathogenesis of PBC.
Evidence of genetic susceptibility to PBCPBC exhibits a number of autoimmune features, including the almost universal presence of autoantibodies reactive with highly conserved mitochondrial antigens. There are geographic and familial clustering of disease, and association with other autoimmune diseases. Patients with PBC are predominantly female. The almost universal presence of such autoimmune features in PBC has led to the view that the disease has an autoimmune pathogenesis.7-9 Like other better characterized autoimmune disease, PBC appears to require genetic susceptibility and a triggering event that initiates the autoimmune attack on bile duct cells.
EpidemiologyRecent studies have suggested that PBC clusters both within geographic areas and families. Clustering of disease within families may involve either shared genes or environment or both. A comprehensive, geographically based cohort study from the North-East of England estimated the sibling relative risk (λs) of PBC at 10.5,10 a value similar to those quoted for other autoimmune disease. The highest risk was found in offspring (λo= 30.6) and particularly daughters of affected mother (λdaughters= 58.7).10 While the ten-fold risk to family members seems to provide strong evidence in support of an inherited basis for PBC, one must be cautious. None of the studies yet performed have managed to distinguish between first degree relatives nutured in a shared environment and those brought up in different environments. New classical approaches to dissecting genetic contribution to complex disease is through the study of concordance rates in twins. Disease concordance rates in monozygotic (who are genetically identical but also share the environmental background) and dizygotic (who share the environmental factors but not the genetic background) twins pairs are powerful tools to estimate the weight of genetic and environmental factors in susceptibility to multifactorial diseases. A more recent report of 16 pairs of twins within a 1,400-family cohort found 5 of 8 monozygotic twin sets in which both individuals had PBC (pair wise concordance rate, 0.63). Among the dizygotic twins, no set was found to be concordant for PBC. Interestingly, the age of onset of disease was similar in 4 of 5 concordant sets of monozygotic pairs; however, there were differences in natural history and disease severity. This data shows not only the role of genetics but also emphasizes that either epigenetic factors and/or environment play a critical role.11
The association with other autoimmune diseasesSome 53 percent of PBC patients have at least one additional autoimmune disease and 37 percent of patients have at least 2 overlapping autoimmune conditions. The most frequently seen associations were with Sjogren’s syndrome (25% of patients), thyroid disease (23%), and rheumatoid arthritis (17%).12 Autoimmunity in the families of PBC patients was common, with 14% of first-degree relatives suffering from autoimmune disease other than PBC.12 This rate is over three times the frequency of autoimmunity in the general population, suggesting a real increased predisposition in these individuals.13 The high incidence of extrahepatic autoimmune disease in PBC patients and their families is both supportive of a heritable component in PBC, suggests that at least some of the genetic risk is non-organ specific, and may be determined by genes that control the nature, expression, and regulation of the immune response in general.
Female predominanceAs observed for most autoimmune diseases, women, especially those of middle age, outnumber men by as much as 10:1 among PBC cases, although such ratio varies widely in different epidemiological studies.14 Given that gender is primarily a genetically determined characteristic, this can be interpreted as evidence in favor of a genetic component to disease susceptibility. The reasons for increased susceptibility in females are not clear.15 In the attempt to explain the female preponderance; the prevailing view is that this gender difference may involve the effects of sex hormones on the immune system. Sex hormones are believed to influence the onset and severity of autoimmune disease by modulating lymphocytes at various stages in life.16 Although specific studies are lacking on the influence of sex hormones on the occurrence of PBC in either sex, such studies have been conducted for other autoimmune conditions, mostly in animal models.17
A considerable number of sex-and immune-related genes have been mapped to the X chromosome; such genes appear crucial in the maintenance of physiological sex hormone levels and, more importantly, of immune tolerance. Monosomy or structural abnormalities of the X chromosome are present in genetic disorders such as Turner’s syndrome18 or premature ovarian failure,19 often presenting accompanying autoimmune features. Finally, various X chromosome disorders can also be accompanied by sign of chronic cholestasis.18 Recent studies show women with PBC manifest an enhanced rate of monosomy X in peripheral blood cells, compared with women with chronic hepatitis C infection or healthy women. The monosomy rates appear to increase with age in all groups and are not influenced by the severity of liver disease or the degree of cholestasis.20 Two possible models can be hypothesized. In the first, haplo-insufficiency of specific X chromosome genes involved in the maintenance of immune tolerance and escape of X inactivation is the key event; secondly, a multigenic complex inheritance model exists in which the Y chromosome exerts a protective effect on susceptibility to PBC.21
The genetic basis of PBCOverall, the above evidence supports the possibility that there is a significant heritable component to PBC. To a geneticist, PBC falls into the category of “complex diseases” characterized by incomplete penetrance, low heritability, and variable phenotypes that often have a strong environmental component. Consequently, understanding the genetic basis of “complex disease” has been heralded as one of the major medical challenges of the post genome era.22
Studies of the genetic basis of disease susceptibility in PBC have been limited. In the absence of the appropriate families in which to use linkage based approaches studies of the genetic basis of PBC performed to date have exclusively used the candidate gene association approach. Given the extensively characterized autoreactive B-and T-cell responses seen in PBC, most studies have focused on genes implicated in the regulation of immune response, and the majority involves the MHC. The only convincingly reproducible association identified to date is with DRB140801 in Northern European populations23-26 and DRB1*0803 in Japanese populations.27-30 Other genetic associations in PBC with chromosome 2q include; cytotoxic lymphocyte antigen-4 (CTL-4),31 interleukin-1 gene cluster,24 caspase 8 gene (casp8),32 and natural resistance associated macrophage antigen 1(nramp1)33 and should be considered with caution until confirmed in dependent series. It is unlikely that the only genes that influence disease susceptibility and progression in PBC are immuno-regulatory genes concerned with T cell immunity. We are a only just beginning to understand the genetic basis of complex disease like PBC. The key issues for future investigation are defining the genetic markers to predict progression and malignancy.
Antimitochondrial antibodiesA major advance in understanding PBC occurred with the identification and cloning of the antigens against anti-mitochondrial antibodies (AMA).34 These antigens constitute the M2 family of mitochondrial antigens, and have been identified as components of the 2-oxo-acid dehydro-genase (2-OADC) family of multi-enzyme complexes; pyruvate dehydrogenase complex (PDC), 2-oxoglutarate dehydrogenase complex (OGDC), and branch-chain oxo-acid dehydrogenase complex (BCOADC)35 which localize to the inner mitochondrial membrane.36 Each catalyzes the reductive transfer of an acetyl group from its respective substrates to coenzyme A for oxidation in the Krebs cycle. Each of the 2-OADC family members has a multimeric structure with multiple substrate-specific decarboxylase dehydrogenase (E1) and dihydrolipoamode dehydrogenase (E3) enzymes attached to a specific multimeric dihydrolipoamode acryltransferase (E2) “core”. PDC has an additional core enzyme E3-binding protein (E3BP), previously termed protein X37(Table I).38-43 The E2 and E1 subunits are specific for the individual complex whilst E3 is common between all 3 family members. It was established that serum autoantibodies from more than 95% of patients with PBC react with PDC-E2 by immunoblotting or enzyme-linked immunosorbent assay (ELISA), whereas the frequency of reactivity against E2 subunit of B CO ADC and OGDC is found in 53% to 88%.38-43 A few AMA-seropositive patients have PDC-E2 reactivity alone, but a majority also showed reactivity against BCOADC or OGDC. Reactivity against BCOADC-E2 or OGDC-E2 alone was less common. The dominant autoepitopes within PDC have been mapped to the E2 and E3BP subunits, with 95% of PBC sera showing reactivity to both antigens.44-45 The seemingly universal co-expression of autoantibody responses reactive to PDC-E2 and E3BP in PBC patients is explained by the complete cross-reactivity between the two antigens resulting from a shared structural motif of an attached lipoic acid moiety.46 The importance of the role played by the lipoic acid cofactor in the immunogenicity of PDC-E2 and E3BP is the dominant autoepitopes within each of these self-antigens.46,47 Recent observations link covalent modification of lipoic acid, and/or exposure to antigenic structures mimicking it, in the initial breakdown of immune tolerance to PDC.48,49
Antimitochondrial autoantibodies and mitochondrial antigen in PBC.
| Antigen | Frequency (%)* | B-cell epitopes | Major Ig isotype |
|---|---|---|---|
| PDC-E2 | 95 | Outer and inner lipoyl domain | IgG3, IgM |
| BCOADC-E2 | 53-55 | Lipoyl domain | NR |
| OGDC-E2 | 39-88 | Lipoyl domain | IgG2, IgM |
| E3BP | 95 | NR | NR |
Although AMA is considered the hallmark diagnostic feature of PBC, it is not the only disease-specific autoantibody. There are several autoantibodies detectable in PBC patients in addition to AMA, some of which appear to be over-represented in AMA-negative PBC. Among them, antinuclear antibody (ANA) is a consistent finding. Two distinct immunofluorescence patterns characterize PBC-specific ANA, namely the perinuclear (rim-like)50 and the multiple nuclear dot (MND) patterns.51 The antibodies responsible for the rimlike pattern recognize constituents of the nuclear envelope, including gp210 and nucleoprotein p62 that may be associated with more active and severe disease.52 Antibodies directed against gp120 are found in about 25% of patients with AMA-positive PBC and up to 50% of those with AMA-negative PBC. The disease specificity for the detection of such autoantibodies by immunoblotting is over 99%.53 The nucleoprotein p62 is a nuclear pore glycoprotein specifically recognized by PBC sera and present in about 25% of patients with PBC.54 The antibodies responsible for the MND pattern recognize the sp100 and promyelocytic leukemia (PML) patterns. Anti-sp100 seropositivity was associated with more frequent progression from early (I/II) to late (III/IV) histological stages over a 24-month observation period.55 Identification of serologic markers associated with increased risk of liver failure would assist in management of PBC patients. ANA-positive PBC patients have liver failure more than ANA-negative PBC patients (41% vs 25%, p = 0.05). The presence of anti-centromere antibodies was associated with liver failure (anti-centromere antibody positive vs. negative, 58% vs. 33%, p = .001).56 PBC patients with ANA may be candidates for treatment with experimental therapies to prolong the interval between diagnosis and liver failure. ANAnegative patients, who appear to have a relatively benign clinical course, should perhaps be treated with ursodeoxycholic acid alone.56 A variety of other auto-antibody targets have been described in PBC, including carbonic anhydrase II,57-59 a-enolase,60 lactoferrin,61 platelet glycoprotein IIa/IIIb,62 smooth muscle antigens, soluble liver antigen/liver pancreas (SLA/LP),63 and the putative transcription factor SCX 13,64 but their clinical significance remain to be established. It would be of interest to determine whether they share motifs common to the 2-OADC proteins.
Target cell biology in PBCOne of the most important enigmas in PBC is the organ specific autoimmune inflammation targeting BEC despite the expression of the autoantigens throughout the entire body. Several lines of evidence have suggested that autoantigens relevant to PBC may be present on the surface of biliary epithelium. The PBC autoantigen PDC is located on the inner surface of the inner mitochondrial membrane and is therefore normally separated from the extra-cellular immune system by three membranes. Monoclonal antibodies to BCOADC-E2 and OGDC-E2, or a molecule that cross-reacts with the inner lipoyl domain of all three enzymes, also show a uniquely intense staining pattern in the apical region of BECs in patients with PBC.65 This surface expression of PDC appears to start early in the disease process occurring before the well characterized up-regulation of MHC class II and intercellular-adhesion molecule-1 (ICAM-1).66 It does not appear to occur as a simple consequence of increased PDC component transcription as an elevation in PDC component mRNA levels is not seen in affected BECs.67 BECs, in contradistinction to other cell types, have been demonstrated to retain immunogenic PDC-E2 while undergoing apoptosis.68 Caspase cleavage of PDC-E2 in vitro has been shown to generate potentially immunogenic protein fragments.69 There is evidence to support the view that the mechanism leading to progressive destruction of bile duct is apoptosis.70 Markers suggestive of the presence of ongoing apoptosis within affected portal tracts include DNA fragmentation and Fas, Lewis Y antigen, bax and bcl-x expression.70-74 Down-regulation of the inhibitor of apoptosis BCL-2 has also been reported.75
Recent data support the hypothesis that apoptotic mechanisms provide an explanation for the abnormal expression of the inner mitochondrial enzyme, PDC, observed on the surface of BECs in the patients with PBC. Using murine and human cell lines, it was found that the induction of apoptosis led to serial release of PDC to cytoplasm and then to the external surface of the plasma membrane providing direct evidence that the antigen on the cell surface is from mitochondrial origin.76 It is interesting to speculate about the implications for PBC of the export of autoantigenic PDC from the inner mitochondrial compartment, where it is concealed from the immune system by 3 membranes, to the external surface of the plasma membrane.77 While the consequences of this redistribution are not known, it is possible that the plasma membrane expressed protein will be bound by cross-reactive anti-PDC antibodies which are produced during some infections.78
The prominent role played by apoptosis in the model may explain disease recurrence in MHC mismatched liver transplant recipients. In this regard, it is intriguing to note the increased rate of disease recurrence reported in patients receiving primary immunosuppressive therapy in the form of tacrolimus compared with patients receiving cyclosporine.79 Cyclosporine has the additional property, not shared with tacrolimus of blocking the mitochondrial permeability transition rendering cells, to some extent at least, resistant to apoptosis.80
Recent data also suggests that the abnormal staining on the BECs is secondary to immune complexes of IgA and IgG coupled to PDC-E2 because of the poly Ig receptor.81 The existence of anti-PDC-E2 IgA in the bile of patients with PBC is also intriguing.82-83 IgA is produced locally within the liver and transported to the lumen by way of a secretory component found only on the BECs.84 The autoreactive IgA may be transported into the BECs and bind PDC-E2 before it reaches the mitochondrial receptor, and this immune complex may serve as source of antigen presented on the surface of the cell. Perhaps this is another role for mucosal bacterial infections which tend to occur with increased frequency in PBC patients.
There is a growing consensus that the specificity of tissue damage in PBC is a result of the unique microenvironment of the biliary tree. BECs are exposed to highly unusual and potentially toxic environment containing both agents excreted in the bile (e.g. xenobiotics) which are able to modify PDC-E2 or heavy metals able to induce apoptosis85 and ascending infectious agents (bacterial or conceivably retroviral agents). Similarly, age related exposure patterns (e.g. the encountering of bioactive compounds such as drugs not typically given to children or work-place toxins) or cumulative exposure resulting in toxic levels being achieved only after many years might explain the late age of onset of PBC.77
Cellular immune response in PBCThe particular specificity of bile duct destruction, the presence of lymphoid infiltration in the portal tracts and the aberrant expression of MHC class II antigen on biliary epithelium in PBC have suggested that an intense autoimmune response is directed against BECs. It has been hypothesized that the destruction of biliary cells in PBC is mediated by autoreactive liver infiltrating T cells through either cytotoxicity or lymphokine production. The identification and characterization of autoreactive T lymphocyte responses is an important step in defining the role of these cells in disease pathogenesis. The evidence was initially derived from histological staining studies of tissue samples and later by analyzing T-cell lines that proliferate in the presence of putative mitochondrial autoantigens. In study by Meuer and colleagues,86 phenotypic and functional analysis of T cells isolated from the liver of patients with PBC demonstrated a heterogenous cell population; however when compared to peripheral blood lymphocytes of the same patients, there was a marked enrichment of CD8+ cytotoxic T cells among the liver T cells. Subsequently, Van de Water and colleagues87 successfully obtained T cell lines from liver biopsies of PBC patients and demonstrated for the first time that clones reacting with PDC-E2 were present in the liver of patients with PBC. In addition, these T-cells were derived from the liver and produced lymphokines responsive to those antigens, including interleukin (IL)-2, IL-4 and interferon-ã suggested that CD4+T cells were present in the liver.
CD4+T cells reactive with PDC-E2 are also present in the peripheral blood mononuclear cell (PBMC) pool in the majority of PBC patients, but almost entirely absent from controls.87-90 Of the T-cell clones derived, several were phenotypically of CD4+ and CD8+ lineage; however, the vast majority were CD4+, consistent with those derived from patients late in the disease when CD4+T cells predominate.87 Epitope mapping studies indicated that 54% of patients had cells that proliferated to the outer lipoyl domain while 36% of patients had cells that proliferated to the inner lipoyl domain.91Shimoda and colleagues92 demonstrated, specifically in PBC, a high precursor frequency of PDC-E2 163-176 reactive T cells in the hilar lymph nodes and liver of patients that was 100150 fold greater than for PBMC from the same patients. Interestingly, autoreactive T cells and autoantibodies from PBC patients both recognize the same immunodominant epitope, which is not always the case for autoimmune disease. The HLA restriction molecules for this epitope were all identified as HLA DRB4*0101.92
Autoreactive CD8+T cells are thought to be involved in the pathogenesis of several autoimmune diseases. Previous investigations have demonstrated the accumulation of antigen reactive CD8+T cells at the site of the inflammation in several autoimmune diseases.93-96 Identification of the autoantigen peptide, that is the target of MHC Class I restricted cytotoxic T lymphocytes (CTLs) in PBC is an important initial step in determining the mechanism by which tolerance to the self peptide is abrogated. Kita and colleagues97 have characterized the first MHC Class I restricted epitope for PDC-E2, namely amino acid 159-167, a region very similar to the epitope recognized by MHC Class II restricted CD4+T cells and antibodies. This PDC-E2159-167 induces specific MHC Class I restricted CD8+ CTL lines from HLA-A2+ PBC patients, but not from controls, following in vitro stimulation of peripheral blood lymphocytes with antigen pulsed-dendritic cells. Furthermore, they also found a 10-fold increase in the frequency of PDC-E2159-167 specific CTLs in the liver as compare to the peripheral blood and in the tissue of patients with early disease.98 These data, for the first time, document the enrichment of autoantigen-specific CD8+ T-cells in the PBC liver, suggesting that CD8+ T-cells play a significant role in the immunopathogenesis of PBC.
Study of other potential cytotoxic effector cells in the liver has so far been limited. Natural killer T (NKT) cells are a subset of lymphocytes incriminated in playing an important role in the modulation of the innate immune response and the development of autoimmunity. However, there have been only limited studies attempting to quantitate the number of NKT cells in autoimmune disease including PBC. An environment of functional NKT cells, quantified by the use of tetramer technology, has been reported in the liver of PBC patients.99 The precise role of NKT cells in the development of PBC remains an enigma.
Dendritic cells (DCs) are professional antigen presenting cells, which are also likely to be important for the induction of immunological tolerance and for the regulation of immune responses. An immunohistochemical study showed increased number of inter-digitating cells as well as highly restricted distribution of CD83+ DCs in PBC, suggesting the role of DCs in PBC.100 PDC-E2 specific CD4+T-cells are induced more efficiently by using antigen pulsed DCs.101 Professional antigen presenting cells (APCs) such as DCs, macrophages, and B cells are capable of intracellular processing of exogenous antigen to generate peptides presented by MHC class I.102,103 The exogenous antigen is presented by the same APCs to CD4+ and CD8+T-cells.104 DCs have been shown to mediate internalization of antigen-immunoglobulin complexes and promote efficient MHC class I as well as class II restricted antigen presentation.105 The maintenance and amplification of CTL response against PDC-E2 with PDC-E2 specific autoantibodies play a key role; i.e. PDC-E2 specific autoantibodies may capture PDC-E2 antigens released from dying cells to form PDC-E2 immune complexes, which may facilitate the uptake of PDC-E2 antigen through Fc y receptors on professional APCs.
Humoral immune response in PBCThe current pathogenic view of PBC is that cellular immunity plays the central role in the development of the disease. The immunodominant antigenic regions recognized by T lymphocytes on PDC-E2 have been defined. These comprise a region targeted by both helper CD4+ and cytotoxic CD8+ T-cells.90,97 The same region is also a target of the receptors of B lymphocytes, which in their soluble form are antibodies. It is conceivable that the distinct arms of the immune system, helper T cells, cytotoxic T cells, and B cells act in concert in inflicting damage to the liver ductules in the pathogenic process of PBC. In support of this view is the recent finding that soluble PDC-E2 complex with a PDC-E2 specific human monoclonal antibody favors generation of PDC-E2 specific cytotoxic cells at a 100-fold lower concentration than otherwise required in the presence of the soluble antigen alone.97 Thus AMA may indirectly play a pathogenic role in facilitating the development of a PDC-E2 specific CD8 T-cell immune responses.
The strength of the association between AMA and the PBC disease process is indicated by the fact that the majority of individuals who are AMA positive in the absence of either symptoms suggestive of PBC or cholestatic liver biochemistry still have features suggestive of early PBC on liver biopsy and, when followed up over a number of years, go on to develop typical disease.106 However, there are no data to support a direct pathogenetic role for AMA. In the setting of human bacterial infection, self-limiting anti-PDC antibody responses can be induced in the apparent absence of other feature of PBC.107 There are no convincing data to support an association between the titer of AMA and nature and extent of bile duct damage.108 Moreover; AMA profiles (A-D) do not predict prognosis in patients with PBC.109
Cytokines in PBCThere are only a few reports dealing with cytokine profiles in patient with PBC. Demonstration of PDC-specific autoreactive T cells in PBC patients and the predominant release of type 1-specific cytokines (IL-2, IFN-γ, and TNF-α) but not type 2-specific cytokines (IL-4, IL-5, and IL-10) support the view that PBC is a delayed-type hypersensitivity-mediated disease. Increase in IL-2, IL-5, IL-6, IFN-γ and transforming growth factor-β mRNA have all been reported in the liver tissue of patients with PBC.110 While this cytokine profile is not specific for PBC, it reflects the inflammatory microenvironment. However, unlike autoimmune hepatitis, there is a preferential upregulation of IL-5 mRNA (a T helper 2-type cytokine gene) in the liver tissues of PBC patients.110
The prototype T helper (Th) 2 cytokines has been proposed to play a role in the establishment and persistence of inflammation in a variety of autoimmune disorders.111 Although the finding of some Th2 prototype cytokines suggests that PBC is also associated with predominantly a Th2 cytokine, the frequency of IFN-γ (a predominant Th1 cytokine) expressing cells predominantly in association with damaged bile ducts provides suggestive evidence that the Th1/Th2 dichotomy in PBC is not clear. In the serum of patients with PBC, levels of IL-5 and IFN-γ are increased.112 Given that eosinophils, growth and differentiation are stimulated by T cell derived cytokines including IL-3, IL-5, and granulocyte macrophage colony stimulating factor and that these cells are found in early disease. It may be that during the progression of PBC, there is a switch from Th2 to Th1. Furthermore, the high levels of IL-10 found in normal but not PBC liver suggest that complex networks of cytokines are involved in regulating the development of liver cell damage and chronic persistent inflammation, and the variability could be associated with disease stage. Recent investigations have examined the role of serum cytokines in PBC, demonstrating the presence of increased levels of TNF-α, IL-8, and IL-12 in advanced compared to early histologic stages.113 The correlation between these findings and disease pathogenesis remain unknown.
Complement System in PBCThe complement system is chronically activated in patients with PBC. Serum concentrations of several complement components are increased. Some patients have circulating immune complexes that result from either increased production or defective clearance by the reticuloendothelial system. These immune complexes may contain antigens that are similar to or cross-reacting with epithelial antigens of bile ducts or mitochondria.114 A C4 abnormality found in patients with PBC115,116 may contribute to deposition of immune complexes or to defective viral clearance, thereby leading to chronic infection possibly triggering autoimmunity. Suppressor T cell activity is abnormal in patients with PBC,117 and in a substantial proportion of their healthy first-degree relatives. This abnormality may be responsible for the increased number of circulating B cells capable of producing AMA spontaneously in vivo.117,118
Oxidative stress in PBCThe pathophysiological mechanisms underlying progression from the initial autoimmune attack to the development of fibrosis and, ultimately, cirrhosis and liver failure in PBC are poorly understood, but recent studies indicates that generation of reactive oxygen species (ROS) such as the superoxide anion and the hydroxyl radical may play a role. ROS were generated by inflammatory processes and possibly by endotoxin or bile acids. Inflammation is a common and early feature of PBC and invading macrophages will generate ROS leading to lipid peroxidation.119 Cholestasis, also a major feature of PBC, leads to accumulation of bile acids and hepatic copper, both of which can lead to generation of free radicals.120,121 One histological study in liver biopsies from five PBC patients demonstrated the presence of malondialdehide (MDA) and 4-hydroxynonenal (HNE) adducts, major metabolites of lipid peroxidation.122 In a larger study, HNE protein adducts have been detected in hepatocytes preferentially located around the portal tracts as well as in the cytoplasm of damaged, but also intact, biliary cells.123 Elevated levels of Mn-superoxide dismutase have been found, and immunostaining of liver biopsies has shown increased expression of this protein in damaged epithelial cells of interlobular bile ducts, bile ductules and degenerated hepatocytes. This finding suggests that free radicals, including the superoxide anion, may be involved in the pathogenesis of PBC.124
Immunohistochemical studies demonstrated the presence of enhanced lipid peroxidation in the bile ducts and hepatocytes of PBC patients.122,123 Pemberton and colleagues125 have assessed a broad spectrum of markers reflecting different facets of the disease process and oxidative and antioxidant activity to support those immuno-histochemical findings. Lipid peroxidation markers were significantly elevated, including plasma and urinary 8-isoprostane and plasma MDA, compared to age-and sex-matched controls. The most striking antioxidant depletion occurred with plasma total glutathione where levels were significantly reduced. Total serum antioxidant levels were decreased and serum selenium and vitamin A were also lower; vitamin C and E were normal. Most patients had early disease biochemically and were Child-Pugh grade A. Urinary 8-isoprostane correlated positively with Ludwig stage and markers of hepatic injury and cholestasis. This study clearly demonstrates that oxidant stress, as reflected in a comprehensive spectrum of lipid peroxidation and antioxidant markers, is a significant feature of early stage PBC.
Previous studies have suggested that increased nitric oxide (NO)-mediated products are found in the livers of subjects with PBC,126,127 but the mechanisms involved remain enigmatic. Wu and colleagues128 examined liver specimens from 64 PBC patients. Twenty two percent of PBC biopsies had elevated expression of 3-nitrotyrosine (the enzymes responsible for NO production) in their BECs. Furthermore, the BECs in PBC also demonstrated apoptotic changes. Myeloperoxidase (MPO)-positive inflammatory cells, the inflammatory cells responsible for the generation of ROS such as O2, H2O2 and NO, were also noted adjacent to the basement membrane. The liver sections of subject with stage I or stage II PBC demonstrated significantly increased inflammatory cell infiltration and elevated 3-nitrotyrosine expression in BECs compared with stage III and IV. The presence of 3-nitrotyrosine was closely associated with infiltrating CD68 and/or MPO positive cells. There was also a stage-associated difference in the presence of bile duct infiltrating cells and 3-nitrotyrosine in PBC with the increase dominant in early stage disease. This study demonstrates NO and ROS, collectively determined as 3-nitrotyrosine, are associated with bile duct destruction in PBC and are particularly prevalent in early stage disease. Therefore, it is still unclear whether oxidant stress is involved in the initiation of the disease process or in its progression.
Hepatic stellate cells (HSCs) play a key role in the development of fibrosis, being a major source of collagen and also of other extracellular matrix proteins in the injured liver. It has previously been shown that stimulation of lipid peroxidation or treatment with HNE increases procollagen mRNA expression in human HSCs129 leading to fibrogenesis and eventually to cirrhosis. Therefore, oxidant stress in PBC provides a likely mechanism for disease progression following the initial immunological insult and leading to the subsequent development of fibrosis and cirrhosis.125
Mechanism of tolerance breakdownThe key pathogenetic step for the development of PBC appears to be breakdown of immune self-tolerance to the highly conserved mitochondrial self-antigen PDC;36 a concept supported by the evidence outlined above implicating autoreactive cytotoxic T-cells as the probable effector cells responsible for BEC killing. The principal B-cell epitope within PDC has been localized to a lipoic acid binding domain within PDC-E2; a key part of the epitope.47 Despite their almost ubiquitous presence in PBC, the role played by PDC-E2 specific antibodies in the pathogenesis of PBC remains unclear. There is an increasing body of evidence to suggest that the effector mechanism responsible for BECs damage in PBC may be apoptosis induced by self-PDC specific cytotoxic effector T cells known to be present at relatively high levels in the peribiliary portal tract infiltrate when BECs damage is known to be occurring.97,98,130 The apparent importance of the increasingly well-characterized autoreactive CD4+ and CD8+ PDC-specific T cell response in the pathogenesis of PBC has led to the obvious question as to how T-cell tolerance breaks down to such a highly conserved and ubiquitously expressed self-antigen as PDC.77 Animal modeling studies suggest that simple exposure to homologues of self-PDC results in breakdown of B-cell but not T-cell tolerance. A reformulation of the key question regarding the pathogenesis of PBC would therefore be: How does breakdown of B-cell tolerance to PDC (an apparently relatively easily achievable state with limited pathological sequelae) result in the much more restricted (and seemingly pathologically significant) state of T-cell tolerance breakdown?
Jones and colleagues131 demonstrated that female SJL/J mice sensitized with foreign PDC rapidly develop high titer autoantibody responses reactive with self-PDC. The development of anti-PDC autoantibodies is not accompanied by breakdown of T-cell tolerance. However, when animals are cosensitized with foreign PDC and self-PDC, autoreactive T-cell responses to self-PDC are seen. The recent data from the same group support the hypothesis that breakdown of T-cell tolerance to PDC can be driven by the presence of B-cells and/or antibodies cross reactive with this self-antigen.132 In this study, they demonstrated that naïve female SJL/J animals break T-cell tolerance to self-PDC when exposed to self-PDC mixed with B cells expressing surface immunoglobulin (Ig) reactive with self-PDC. Such tolerance breakdown was absent from animals exposed to either self-PDC or self-PDC reactive B cells in isolation, or from animals exposed to self-PDC mixed with control B cells. These data suggest that the B-cell response to self-PDC so characteristic of PBC may play a critical role in driving the PDC-specific autoreactive T-cell response implicated in target cell damage. There are two potential mechanisms for antigen-specific B-cell promoted breakdown of T-cell tolerance to self-PDC.132 The first potential mechanism is through binding of self-PDC to reactive Ig B-cell surface receptors increasing uptake, processing, and presentation of self-derived epitopes.133 It has been demonstrated that B cells act as professional antigen-presenting cell (APC) breaking T-cell tolerance, provided they have an activated phenotype and express surface receptor reactive with the antigen to which T-cell tolerance is broken.134 The second potential mechanism is through self-PDC reactive antibody-promoted uptake and presentation of self-antigen by other APCs through the formation of immune complexes taken up and processed at an increased rate by APCs.135
These observations would be compatible with a two-stage model for disease pathogenesis. In the first stage, an antibody response reactive with self-PDC would be induced. The induction of this response would be necessary, but in isolation insufficient for the induction of PBC. In the second stage, this B-cell response would drive the breakdown of T-cell tolerance seemingly associated with target cell damage. Important questions remain regarding the second stage of this model. These include the source of the self-PDC needed to drive T-cell tolerance breakdown in the context of cross-reactive B cells and the stimuli required to maintain a state of B-cell activation.132
With regard to the first stage of the model, human and animal studies have suggested that the induction of an antibody response reactive with self-PDC may result from a number of different priming events. Among the events demonstrated to induce an antibody response cross-reactive with self-PDC are exposure to bacterial PDC78,136 or other microbial structure mimics,137 retroviral proteins,138 and exposure to chemically modified PDC or mimicking xenobiotics48,49 or microchimerism.139 The diversity of the potential events giving rise to antibody responses cross-reactive with PDC, which could promote subsequent T-cell tolerance breakdown, suggests the intriguing possibility that PBC could represent a condition with a common final pathway but with multiple triggers able to induce a B-cell response cross-reactive with self-PDC.132
Molecular mimicryMolecular mimicry for an extrinsic protein of an infectious agent has long been suggested as a possible initiating event in PBC. Molecular mimicry between host autoantigens and unrelated exogenous protein is one of the hypotheses used to explain how autoantibodies to self proteins arise to break tolerance and lead to autoimmune disease. PDC-E2 is a well conserved molecule among various species, especially at the lipoic acid-binding sites. Three conceptual problems arise with such models.77 First, the most prevalent ‘mimic’ of self-PDC is obviously bacterial PDC which is immunogenic but not, it would appear, pathogenic. Secondly, although studies demonstrating reactivity with epitopes derived from pathogen genetic sequences are tantalizing, they fail to show whether such potentially cross-reactive epitopes are generated in vivo during natural infection. Finally, a recent study addressing changes in specificity of human anti-PDC antibodies during affinity maturation argues directly against such molecular mimicry models.140
Despite the lack of evidence to support simple molecular mimicry models, several findings do suggest some role for bacterial infection with increased prevalence of bacterial infection in PBC patients,141,142 serological evidence of specific previous infection143 and bacterial products being present in the mononuclear cells surrounding damage interlobular bile ducts.144 Features of the host response seen in PBC such as the presence of monocyte chemotactic protein (MCP)-2 and MCP-3 expressing mononuclear cells in the portal tract infiltrate and around the periphery of the archetypal epithelial granulomata have also been interpreted as suggesting a role for localized bacterial infection.145
Infectious agents could contribute to the pathogenesis of PBC in one of two ways; as direct pathogenetic factors (infecting BEC and directly inducing apoptosis or acting as a stimulus for immune mediated clearance of infected cells) or as triggers for autoimmunity. Several candidate organisms have been proposed as triggers of PBC but clear causal relationship has not been established. There is cross reactivity between AMA and some of the enzymes involved in oxidative energy metabolism in microorganisms such as E.coli, Lactobacillus delbrueckii, and Chlamydia spp.146-148 However, it is uncertain that any of these microorganisms has a causal role in the development of PBC since antibody titers against these bacterial enzymes are significantly lower than titers of human AMA in PBC patients. Furthermore, these antibodies appear later in the course of PBC.149
Emerging data suggest an etiologic role for another bacterium, Novosphingobium aromaticivorans, a gram negative, strictly aerobic bacteria that is found worldwide in soil, water and coastal plain sediments.150 PDC-E2 like proteins from this bacterium have a higher degree of homology with the immunodominant region of human PDC-E2 than with any microorganism thus far studied (10-1,000 times greater than that of E.coli).151 In an initial study, human PBC sera had titers to N. aromaticivorans protein that were similar to those to human PDC-E2.151 In addition, N. aromaticivorans can metabolize chemical compounds that are similar to the xenobiotics that are reactive against sera from PBC patients.152
Recent study showed PBC patients who had antibodies to PDC-E2 and reacted to lipoylated proteins from N. aromaticivorans, in titers up to 1:1,000,000.151 Twelve percent of AMA-negative PBC patients also reacted against those proteins, suggesting that the close homology between bacterial and human PDC-E2 alone does not explain the reactivity of PBC sera to the N. aromaticivorans proteins. Sera from the control group were negative. Of interest, one healthy first-degree relative had antibodies to N. aromaticivorans and developed PBC 2 years later. In addition, approximately 25% of PBC patients and controls had N. aromaticivorans in their stools but only those with PBC had antibodies to N. aromaticivorans. The clinical implications of these data are uncertain. There are many important questions such as whether some PBC patients are persistent carriers of N. aromaticivorans, and whether testing for exposure and treatment with antibiotics might alter the natural history of the disease or prevent its development in patients at risk. A more detailed understanding of the epidemiology and immuno-pathogenesis is needed.153
Retroviral InfectionAn alternative model for the pathogenesis of PBC invokes retroviral infection as a key etiological process. Preliminary data from one laboratory suggest that infection with a retrovirus may be associated with PBC in some patients.138,154,155 Electron microscopy, cloning and immunohistochemistry have shown a viral particle that has been tentatively identified as a human beta retrovirus. Viral proteins colocalized with cells demonstrating aberrant autoantigen expression. Furthermore, lymph node homogenates from patients with PBC induced autoantigen expression in normal biliary epithelial cells, which also developed phenotypic manifestations of PBC. Although intriguing, this latter observation does not show a clear causality between the beta-retrovirus and the development of PBC or the expression of PDC-E2 like antigen on the cell membrane. At a general level, the role played by retroviral infection in the pathogenesis of autoimmunity has been much debated and several etiological agents have been identified in different autoimmune diseases. These ret-roviral associations have usually not stood up to closer scrutiny and the whole question of the role played by retrovirus in autoimmune disease remains controversial.103
XenobioticsOther environmental factors have received growing attention as possible triggers of autoimmunity in PBC. Xe-nobiotics are foreign compounds that may alter defined self protein, inducing a change in the molecular structure of the native protein sufficient to induce an immune response. Such immune responses may then result in the recognition of not only the modified protein, but also the native form.156,157 The chronic presence of the self protein serves to perpetuate the immune response initiated by the xenobiotic-induced adduct thus leading to chronic autoimmunity.158,159 Many xenobiotics are metabolized in the liver, thereby increasing the potential for liver specific alteration of protein.160
To address the hypothesis that PBC is induced by xenobiotic exposure, Long and colleagues152 synthesized the inner lipoyl domain of PDC-E2, replacing the lipoic acid moiety with synthetic structures, and quantitated the reactivity of these structures with sera from PBC patients. Results showed that AMA from all seropositive patients with PBC, but not from controls, reacted against 3 of the 18 organic modified autoepitopes significantly better than to the native domain. As many chemicals, including pharmaceuticals and household detergents, have the potential to form such halogenated derivatives as metabolites, such findings indicated for the first time that an organic compound may serve as a mimotope for an autoantigen, thus further providing evidence for a potential mechanism by which environmental organic compounds may cause PBC. It was demonstrated that one of these compounds is 6-bromohexanoate, conjugated with a protein backbone unrelated to PDC-E2, elicited a specific immunogenic response with AMA production in animal models.48 This observation indicates that lipoic acid residues and possibly some xenobiotic-derived variants are crucial in the epitope recognition. The lack of liver lesions in these models could be accounted for by the natural course of PBC observed in humans, with AMA appearing long before the clinical manifestation of PBC.34
MicrochimerismMicrochimerism is the presence of a low level of nonhost stem cells or their progeny in an individual. The most common source of microchimerism is pregnancy. During pregnancy, bi-directional trafficking of hemato-poietic cells occurs through the placenta and these micro-chimeric cells persist long after pregnancy.161 The question of the association between fetal microchimerism and PBC has been addressed in recent studies.162-165 These studies have all used molecular biology approaches to detect the presence of Y-chromosome derived sequences in the peripheral blood and/or liver tissue of female PBC patients. In all the studies, significant numbers of PBC patients lacked evidence of microchimerism. Moreover, the disease clearly occurs in both men and nulliparous women in whom the process cannot have occurred. Tellingly, the numbers of pregnancies reported by PBC patients also appears to be the same as for controls in a reported case control study,166 suggesting no association between numbers of pregnancies and disease. Conversely, the fact that fetal microchimerism is seen in a population of normal individuals suggests that the process is not, in isolation, sufficient to induce the disease. Presumably, any susceptibility to the induction of PBC resulting from the presence of fetal microchimerism results in expression of the disease only in individuals exposed to other additional environmental or genetic risk factors.15 Intriguingly, Corpechot and colleagues161 showed that the prevalence of antinuclear antibody (ANA) is significantly higher in PBC patients showing evidence of fetal microchimerism. Moreover, the presence of anti-centromere antibody (ACA), which shows a significant association/overlap with PBC,167,168 was also associated with the presence of fetal micro-chimerism. The implication of this observation is that fetal microchimerism may act as a trigger for disease in the subgroup of patients with ANA and ACA.
Primary biliary cirrhosis: the futurePrimary biliary cirrhosis is a disease of modern times. The first description of PBC occurred in 1857 by Addison and Gull, and advances in the study of the pathogenesis of PBC have occurred over the last 20 years. The direction of research has been heavily influenced by the observation that patients typically have high titers of serum autoanti-bodies directed against highly conserved autoantigens (mitochondria, most classically pyruvate dehydrogenase complex). Major advances in communication technology have facilitated worldwide awareness of this disease and rapid exchange of scientific information. Although the study of PBC is significantly impeded by the lack of an adequate animal model, the molecular technology to address these issues is advancing rapidly. These advances can be anticipated to accelerate the pace of new discoveries in the area of the pathogenesis of PBC to improving the diagnosis and treatment of PBC patients. Several critical questions can be posed as priorities for research in the future.
- 1.
Which genes are important in the pathophysiology of PBC?
- 2.
Why does PBC primarily affect women?
- 3.
What specific role to anti-PDC responses play in target cell damage?
- 4.
How and why does immune tolerance breakdown to an antigen present in all nucleated cells result in damaged restricted primarily to the intrahepatic bile ducts?
- 5.
How and why does the disease recur following liver transplant?
Primary biliary cirrhosis is a chronic, progressive cholestatic liver disease of unknown cause with the presence of a serologic hallmark, namely the antimitochondrial antibody. It’s predilection for women and its association with other autoimmune disease suggests an immune based pathogenesis. Current theories on the etiopathogenesis of PBC favor the hypothesis that the disease develops as a result of an inappropriate (genetically controlled) immune response following stimulation by an environmental or infectious agent. The pathogenetic mechanism is believed to be caused by a defect in immunologic tolerance, resulting in the activation and expansion of self-antigen specific T and B lymphocyte clones and the production of circulating autoantibodies in addition to a myriad of cytokines and other inflammatory mediators. Furthermore, apoptotic BEC have been found in the PBC liver, which is consistent with this mode of tissue injury. The primary events are assumed to be specific for the target tissue, which are further enhanced by a secondary bystander response to cellular components released from dying cells. The biggest impedance to improving treatment of patients remains its elusive pathogenesis. Several critical questions can be posed as priorities for researchers in the future. Which genes are important in the pathophysiology of PBC? Why does PBC primarily affect women? What specific role to anti-PDC responses play in target cell damage? How and why does immune tolerance breakdown to an antigen present in all nucleated cells result in damaged restricted primarily to the intrahepatic bile ducts? How and why does the disease recur following liver transplant? The answers to these questions are keys to understanding the pathogenesis of PBC. Although the study of PBC is significantly impeded by the lack of an adequate animal model, the molecular technology to address these issues is advancing rapidly. These advances can be anticipated to accelerate the pace of new discoveries in the area of the pathogenesis of PBC.



