



In parallel to the human genome sequencing project, several technological platforms have been developed that let us gain insight into the human genome structure, as well as evaluate their usefulness in the clinical approach of patients. Thus, in acute lymphoblastic leukemia (ALL), the most common pediatric malignancy, genomic tools promise to be useful for detecting patients at high risk of relapse, either at diagnosis or during treatment (minimal residual disease), and they also increase the possibility to identify cases at risk of adverse reactions to chemotherapy. Therefore, the physician could offer patient-tailored therapeutic schemes. A clear example of the useful genomic tools is the identification of single nucleotide polymorphisms (SNPs) in the thiomethyl-purine transferase (TPMT) gene, where the presence of two null alleles (homozygous or compound heterozygous) indicates the need to reduce the dose of mercaptopurine by up to 90% to avoid toxic effects which could lead to the death of the patient.
In this review, we provide an overview of the genomic perspective of ALL, describing some strategies that contribute to the identification of biomarkers with potential clinical application.
En paralelo al proyecto de la secuenciación del genoma humano, se han desarrollado varias plataformas tecnológicas que están permitiendo ganar conocimiento sobre la estructura del genoma de las entidades humanas, así como evaluar su utilidad en el abordaje clínico del paciente. En la leucemia linfoblástica aguda (LLA), el cáncer infantil más común, las herramientas genómicas prometen ser útiles para detectar a los pacientes con alto riesgo de recaída, ya sea al diagnóstico o durante el tratamiento (enfermedad mínima residual), además de que permiten identificar los casos en riesgo de presentar reacciones adversas a los tratamientos antineoplásicos y ofrecer una medicina personalizada con esquemas terapéuticos diseñados a la medida del paciente. Un ejemplo claro de esto último es la identificación de polimorfismos de un solo nucleótido (SNPs) en el gen de la tiopurina metil transferasa (TPMT), donde la presencia de dos alelos nulos (homocigotos o heterocigotos compuestos) indica la necesidad de reducir la dosis de la mercaptopurina hasta en un 90% para evitar efectos tóxicos que pueden conducir a la muerte del paciente.
En esta revisión se proporciona una visión global de la genómica de la LLA, describiendo algunas estrategias que contribuyen a la identificación de biomarcadores con potencial utilidad en la práctica clínica.
In Mexico, cancer is a public health priority problem due to its incidence and high mortality rate.1,2 Recent estimates indicate that between 2600 and 3120 cases of cancer are annually diagnosed in people younger than 18 years old, becoming the first leading cause of death in children between five to fourteen years old. It has been reported that 25% of these cancers are acute lymphoblastic leukemia (ALL).1–3 Lately, there have been great advances in the treatment of ALL, achieving a cure rate of up 90%, unlike the 10% that was achieved 50 years ago.4–6 On the one hand, this success is due to the scientific and technological development, which has allowed the improvement of antibiotics and has established timely diagnosis, early detection of malignant clones to identify patients at high risk of relapse, the discovery of more efficient anti leukemia drugs and the design of more specific therapeutic schemes with reduced negative side effects. On the other hand, increased knowledge of the biology of ALL is a fundamental basis for today's high remission rates.5–7 To this date, the treatment of ALL is based on the risk of relapse every patient has, which is mainly determined by age and leukocyte blood count (LBC) at diagnosis, infiltration to other organs, immunophenotype and presence of cytogenetic and molecular alterations. Thus, compared to low or standard risk cases, patients at high risk of relapse are treated with more aggressive protocols or innovative therapies trying to achieve higher cure rates.4–7 Parallel to the project of sequencing the human genome, whose first draft was published in April 2001,8,9 various technology platforms have been developed that are increasing knowledge about the structure of ALL genome, the transcriptome of coding and noncoding genes (miRnoma) and the proteome of a tissue in a given moment, and on the genes related to the metabolism of drugs and therapeutic response (pharmacogenomics). Genomic tools promise to be useful in identifying patients at high risk of relapse at diagnosis and during treatment (MRD, minimal residual disease) and to enable the offering of tailored therapeutic regimens for every patient.
This review provides an overview of ALL genomics and of some biomarkers with potential application in clinical practice.
2Clinical diagnosis of ALLALL represents a group of onco-hematological rapidly evolving clinically and biologically heterogeneous entities, characterized by uncontrolled proliferation of immature white blood cells in the bone marrow (BM) and blood and infiltration of these cells into other tissues. The most common symptoms include fever (caused by leukemia or secondary to serious infections if neutropenia is present), fatigue, anemia, bleeding, bone or articular pain, petechiae and ecchymoses.10,11 More severe clinical manifestations include dyspnea, hepatomegaly, splenomegaly, lymph adenopathy, mediastinal and testicular infiltration.7,10,11
Differential diagnosis of ALL is based on cytochemical staining properties (negative myeloperoxidase, Sudan black B, alpha-naphthyl acetate esterase) and immunophenotype of leukemic cells. Until recently, the Cooperative French-American-British (FAB) Group classified ALL based on the morphology of leukemic cells (subtype L1, L2, and L3); however, the actual classification is based on immunophenotype.11–13 Pre-B ALL is mainly characterized by the expression of cytoplasmic immunoglobulins (cIg) and markers such as CD79a, CD19, HLA-DR and CD10; B cells ALL is characterized by the expression of surface immunoglobulins (sIg) and heavy chains μ; finally, T-cell ALL is characterized by the cytoplasmic expression of CD3, CD7, CD5 or CD2. Also, a subgroup of ALL called pre-B transitional is characterized by the expression of cytoplasmic ion heavy chains μ in immunoglobulins, and weak expression of these surface chains, without the presence of light chains λ or κ.10,11 A small group (<5%) of ALL cases have ambiguous lineage since they express lymphoid and myeloid markers (biphenotypic) or have two cell populations (bilinear).10,13,14
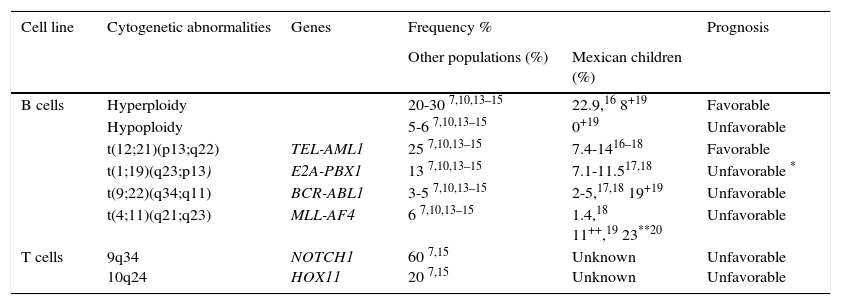
2.1Cytogenetics and molecular geneticsRegarding genetics, ALL is a complex and heterogeneous entity. Aneuploidies (hyperdiploid > 50 chromosomes and hypodiploidy to <44 chromosomes) and chromosomal translocations are the genetic alterations most commonly found in this condition. In pediatric leukemias, B-cell lineage translocations t (12;21) (TEL-AML / ETV6-RUNX1), t (1;19) (E2A-PBX1 / TCF3-PBX1) and t (9;22) (BCR-ABL) and fusions involving the MLL (primarily MLL-AF4) gene are the best characterized and most frequently found genetic abnormalities. On average, ETV6-RUNX1 has been reported in 25% (range 3-45%) of cases, followed by TCF3-PBX1 (13%), BCR - ABL (3-5%) and MLL-AF4 (6.0%).7,10,13–15 In Mexican patients, the first three changes are the most common and constitute between 17.7% to 28.8% of all genetic abnormalities,16–18 although BCR-ABL has been reported in frequencies up to 19% in the population of Veracruz.19 Meanwhile, MLL gene rearrangements have been reported in 1.4% of all cases and in 23% of patients younger than 26 months.18−20 Furthermore, T-cell ALL is characterized by mutations in NOTCH1 (up to 60% of cases) and rearrangements in TLX1-HOX11 (5-10%), TLX3-HOX11L2 (20%), and others (Table 1).7–15
Genetic abnormalities detected by cytogenetic more frequently and ALL molecular biology.
| Cell line | Cytogenetic abnormalities | Genes | Frequency % | Prognosis | |
|---|---|---|---|---|---|
| Other populations (%) | Mexican children (%) | ||||
| B cells | Hyperploidy | 20-30 7,10,13–15 | 22.9,16 8+19 | Favorable | |
| Hypoploidy | 5-6 7,10,13–15 | 0+19 | Unfavorable | ||
| t(12;21)(p13;q22) | TEL-AML1 | 25 7,10,13–15 | 7.4-1416–18 | Favorable | |
| t(1;19)(q23;p13) | E2A-PBX1 | 13 7,10,13–15 | 7.1-11.517,18 | Unfavorable * | |
| t(9;22)(q34;q11) | BCR-ABL1 | 3-5 7,10,13–15 | 2-5,17,18 19+19 | Unfavorable | |
| t(4;11)(q21;q23) | MLL-AF4 | 6 7,10,13–15 | 1.4,18 11++,19 23**20 | Unfavorable | |
| T cells | 9q34 10q24 | NOTCH1 HOX11 | 60 7,15 20 7,15 | Unknown Unknown | Unfavorable Unfavorable |
Knowledge of genetic alterations in ALL has favorably improved, thanks to the constant development of technological tools that enable the rapid analysis of hundreds of genes or the entire genome of hundreds of individuals. In this race of knowledge, the most useful strategies have been DNA microarrays and expression, massive sequencing (genome, exome, transcriptome, among others), and 5’exonuclease.21–27
3.1Parallel or massive sequencingMassive sequencing techniques or next generation sequencing (NGS) can sequence thousands of nucleotides simultaneously in a single reaction, whether covering the entire genome (WES, whole-exome sequencing), or only the exome, the transcriptome (expressed genes) or a panel of selected genes.21 The WES is expensive and can be inefficient in GC-rich regions (mainly promoter regions) but, unlike other approaches, provides the ability to identify structural genetic alterations such as large rearrangements and deletions/insertions. In contrast, the exome sequencing approach has a lower cost than WES and is useful for finding mutations in coding regions. Moreover, since this tool can achieve a wide coverage (200X per haploid genome), it can detect mutations in underrepresented clones, which is very helpful to detect minimal residual disease (MRD) and identify patients with high relapse risk.21,22
Transcriptome sequencing involves the analysis of all expressed genes (gene rearrangements, new isoforms), including antisense and small (microRNAs) and long RNAs (lncRNAs, long non-coding RNA) among others.23 For the detection of all forms of RNA, (expressed or not), the RNA-seq strategy is currently the most popular tool.21,23
3.2DNA and expression microarraysMicroarray platforms have enabled the analysis of structural chromosomal alterations (cytogenetics) that could not be detected with conventional cytogenetic techniques or molecular cytogenetics, including chromosomal translocations, insertions/deletions, duplications, among others.28 They have also contributed to the identification of genetic variants that increase the risk of having leukemia by analyzing thousands of variants of polymorphisms of a single nucleotide (SNP) and variations in the copy number of relevant genes involved in normal and oncogenic hematopoiesis.22,28–30 These approaches are presented without a hypothesis but by comparing cases with controls and generating a global picture of the genetic structure of ALL. With DNA microarrays, which include thousands of cases compared with alleles and controls, it has been possible to identify novel loci associated with ALL; however, a major limitation is that it requires a large sample size to detect alleles of small effect, which is also costly.
The expression of coding and not - coding genes by microarray assays only detect genes represented with probes in the array; however, identifies new genes involved in the etiopathogenesis of ALL, inferring possible signaling pathways and interactions between genes.23 From the clinical standpoint, this knowledge has led to the discovery of various molecular subtypes of which strongly correlate with the prognosis of the disease.25,31–33
3.35’ exonuclease and candidate geneCandidate gene approach techniques part from the hypothesis that certain polymorphisms in genes involved in the pathological processes that characterize leukemia (blast proliferation, lymphopenia, leukopenia, and thrombocytopenia) can contribute to the risk of disease, modify its clinical course or influence its response to treatment. Other genes of interest include those whose variations have evidence of association with leukemia in different populations or have demonstrated its involvement in oncogenesis in animal models. The candidate gene approach compares the frequency in which these polymorphisms occur in a group of affected individuals compared to a group of healthy subjects. If these differences are statistically significant, it is suggested that the analyzed variant is associated with the disease in question. A significant limitation of this approach is that a large group of patients and controls matched by ethnic origin are required to avoid false positive results derived from population stratification. However, it is a strategy that allows identifying genes that have a small effect on the disease, in addition to being more affordable compared to other approaches. The most useful tool for genotyping genomic is the 5’exonuclease technique, which allows the analysis of an SNP in hundreds of samples in less than twohours using specific fluorescent labeling probes.27,34
4Genomics of ALLAlthough chromosomal translocations are markers of ALL, genomic tools have revealed that other highly recurrent alterations are involved in its pathogenesis, evolution, severity, and response to treatment. The nature of these mutations include single base changes of deletions/insertions (indel), duplications and variations in the number of copies (CNVs). The most frequently affected genes are involved in lymphocyte differentiation (PAX5, IZKF1, EBF1 and LMO2), tumor suppressors and cell cycle regulators (DKN2A / CDKN2B, PTEN, and RB1), transcription regulators and co-activators (TBL1XR, ETV6, and ERG), among others. Similarly differences in the frequency of alterations between individuals carrying specific translocations have been reported. For example, in positive BCR-ABL and TEL-AML1 ALL, there are more additional alterations than in ALL with MLL rearrangements. Meanwhile, in hypodiploid ALL, mutations in IKAROS, IKZF2 and genes involved in the RAS pathway are frequently detected. The subtype of ALL without cytogenetic abnormalities is characterized by mutations in ETS transcription factors.22,32,35–41
4.1Associated genesIn ALL, a small number of cases show a Mendelian inheritance pattern, where TP53, PAX5, and GATA2 genes are the most recognized as responsible for an autosomal dominant transmission.42–45 More recently and with the identification of the germinal mutations p. L349P and N385fs in ETV6, it was also observed that ALL might have a recessive inheritance pattern. It is noteworthy that heterozygotes (two different alleles) for these mutations developed entities associated with ALL, such as thrombocytopenia and other solid tumors.46 However, ALL is mainly multifactorial, a complex pattern of inheritance, where polymorphisms in low-penetrance genes involved in DNA repair, in response to xenobiotics, in the immune system and regulatory genes predispose to the development of the disease.47,48 An interesting finding in these studies is the identification of SNPs in intron 3 of ARID5B (AT Rich Interactive Domain 5B) as risk factors for ALL. In fact, in the analyzed populations (including Mexico), this is the most replicated gene.31,48–55ARID5B encodes a member of the family of DNA binding proteins with an interaction domain rich in AT. The protein forms a complex of histone methylases H3K9me2 with two zinc fingers PHD type and is involved in adipogenesis, liver development and differentiation of B – lymphocytes. It has been reported that homozygous carriers of risk alleles not only have a higher odds ratio (OR), but also respond better to treatment with methotrexate.30,49–54
Other widely studied genes are IKZF1, CEBPE, PIP4K2A and CDKN2A / CDKN2B, TP53, and GATA3.29,37,39,40,45,54–58 From the later, variants rs3824662 and rs3781093 confer susceptibility to ALL in children, adolescents and young adults, and are associated with age at diagnosis as well as the presence of rearrangements in CRLF2, mutations in JAK and deletions in IKZF1, genes that directly influence the transcription of GATA3.29,31,38 Also, it has been reported that risk alleles in IKZF and CEBPE distinguish B-cell ALL from T cell ALL.41
Other studies suggest that genes involved metabolism or hydrocarbons which have a potential role in innate and adaptive host immune response may have relevance in the etiology of ALL.47,59–63 In the first group, there are CYP1A1, CYP2D6, CYP2E1 and CYP3A5 as well as XRCC1, GSTP1, GSTT1, CYP2E1, NAT2, and NQO1, which have also been associated with high risk of early relapse, chemotherapy toxicity, reduced resistance to antineoplastic agents and lower survival rate.61,63–65 Some associations depend on the ethnic origin of gene-gene interactions, age and type of leukemia. For example, the CYP1A1*2A allele confers risk for ALL in Caucasian children and is greater if the individual is homozygous for CYP1A1 * 2A and GSTM1 null genotype.63 In the second group, there are IL12A, HLA-DP, HLA-DOA, IL12A, CD28, FCGR2, GATA3, STAT4, and STAT6, among others.62 Chang et al.62 observed that the G allele of rs583911 of IL12A confers an OR of 1.52 for ALL in children regardless of their ethnic origin; however, the CCGA-IL12 haplotype (rs6602398, rs942201, rs791587 and rs706778) increases OR to 9.15 only in children of Hispanic descent. Meanwhile, the haplotype GAC (rs17769459, rs4853546, and rs1031509) of STAT4 has a differential effect risk in Hispanic and non-Hispanic children (OR = 2.65 and 0.30, respectively).
5Ethnical origin as risk factorThe differential contribution of genes and their variants in the etiology of ALL among different ethnic groups is a common finding. Although the population stratification, sample size, are factors that could influence this phenomenon, there is growing evidence that the genetic background of patients, intrinsic to their ethnic origin, constitutes in itself a risk factor for the disease. For example, it has been documented that Native American populations (50.6 cases per million) have a higher prevalence of ALL compared with other populations (Hispanic, 40.9; Caucasian, 35.6, and African American, 14.8 million).66–71 Also, T-cell ALL is up to 1.7 times higher in African Americans than in other populations; the TCF3-PBX1 fusion is also over-represented in this population, while the ETV6-RUNX1 gene is more common in Caucasians than in Hispanics71. Using ancestry genetic markers, it was reported that Native American children have an increased risk of relapse independently of other prognostic factors such as age at diagnosis, LBC, lineage or molecular subtype of leukemia.50
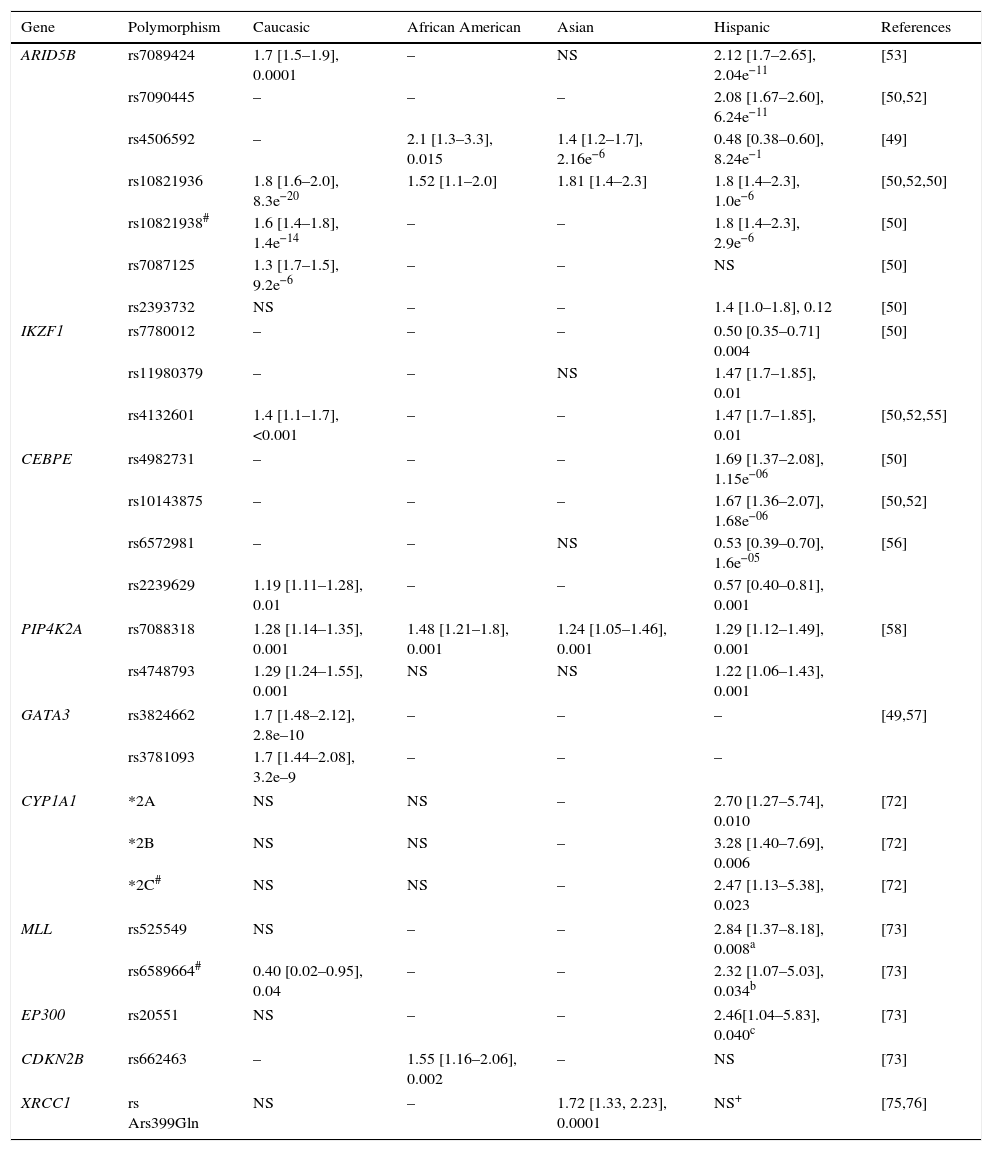
On the other hand, ARID5B genotypes associated with a higher risk of ALL are more common in Hispanic populations compared to other ethnic groups; additionally, specific variants in these populations and for Caucasians have been identified (Table 2).29,31,39,40,45,50,54,55,72–76 The SNP (rs7088318) of PIP4K2A associated with a higher risk for ALL also has a differential distribution in various ethnic groups31,58; therefore, risk genotypes for ALL in ARID5B and PIP4K2A seem to be significant racial determinants for ALL susceptibility.
Example of polymorphisms conferring risk of developing acute lymphoblastic leukemia.
| Gene | Polymorphism | Caucasic | African American | Asian | Hispanic | References |
|---|---|---|---|---|---|---|
| ARID5B | rs7089424 | 1.7 [1.5–1.9], 0.0001 | – | NS | 2.12 [1.7–2.65], 2.04e−11 | [53] |
| rs7090445 | – | – | – | 2.08 [1.67–2.60], 6.24e−11 | [50,52] | |
| rs4506592 | – | 2.1 [1.3–3.3], 0.015 | 1.4 [1.2–1.7], 2.16e−6 | 0.48 [0.38–0.60], 8.24e−1 | [49] | |
| rs10821936 | 1.8 [1.6–2.0], 8.3e−20 | 1.52 [1.1–2.0] | 1.81 [1.4–2.3] | 1.8 [1.4–2.3], 1.0e−6 | [50,52,50] | |
| rs10821938# | 1.6 [1.4–1.8], 1.4e−14 | – | – | 1.8 [1.4–2.3], 2.9e−6 | [50] | |
| rs7087125 | 1.3 [1.7–1.5], 9.2e−6 | – | – | NS | [50] | |
| rs2393732 | NS | – | – | 1.4 [1.0–1.8], 0.12 | [50] | |
| IKZF1 | rs7780012 | – | – | – | 0.50 [0.35–0.71] 0.004 | [50] |
| rs11980379 | – | – | NS | 1.47 [1.7–1.85], 0.01 | ||
| rs4132601 | 1.4 [1.1–1.7], <0.001 | – | – | 1.47 [1.7–1.85], 0.01 | [50,52,55] | |
| CEBPE | rs4982731 | – | – | – | 1.69 [1.37–2.08], 1.15e−06 | [50] |
| rs10143875 | – | – | – | 1.67 [1.36–2.07], 1.68e−06 | [50,52] | |
| rs6572981 | – | – | NS | 0.53 [0.39–0.70], 1.6e−05 | [56] | |
| rs2239629 | 1.19 [1.11–1.28], 0.01 | – | – | 0.57 [0.40–0.81], 0.001 | ||
| PIP4K2A | rs7088318 | 1.28 [1.14–1.35], 0.001 | 1.48 [1.21–1.8], 0.001 | 1.24 [1.05–1.46], 0.001 | 1.29 [1.12–1.49], 0.001 | [58] |
| rs4748793 | 1.29 [1.24–1.55], 0.001 | NS | NS | 1.22 [1.06–1.43], 0.001 | ||
| GATA3 | rs3824662 | 1.7 [1.48–2.12], 2.8e–10 | – | – | – | [49,57] |
| rs3781093 | 1.7 [1.44–2.08], 3.2e–9 | – | – | – | ||
| CYP1A1 | *2A | NS | NS | – | 2.70 [1.27–5.74], 0.010 | [72] |
| *2B | NS | NS | – | 3.28 [1.40–7.69], 0.006 | [72] | |
| *2C# | NS | NS | – | 2.47 [1.13–5.38], 0.023 | [72] | |
| MLL | rs525549 | NS | – | – | 2.84 [1.37–8.18], 0.008a | [73] |
| rs6589664# | 0.40 [0.02–0.95], 0.04 | – | – | 2.32 [1.07–5.03], 0.034b | [73] | |
| EP300 | rs20551 | NS | – | – | 2.46[1.04–5.83], 0.040c | [73] |
| CDKN2B | rs662463 | – | 1.55 [1.16–2.06], 0.002 | – | NS | [73] |
| XRCC1 | rs Ars399Gln | NS | – | 1.72 [1.33, 2.23], 0.0001 | NS+ | [75,76] |
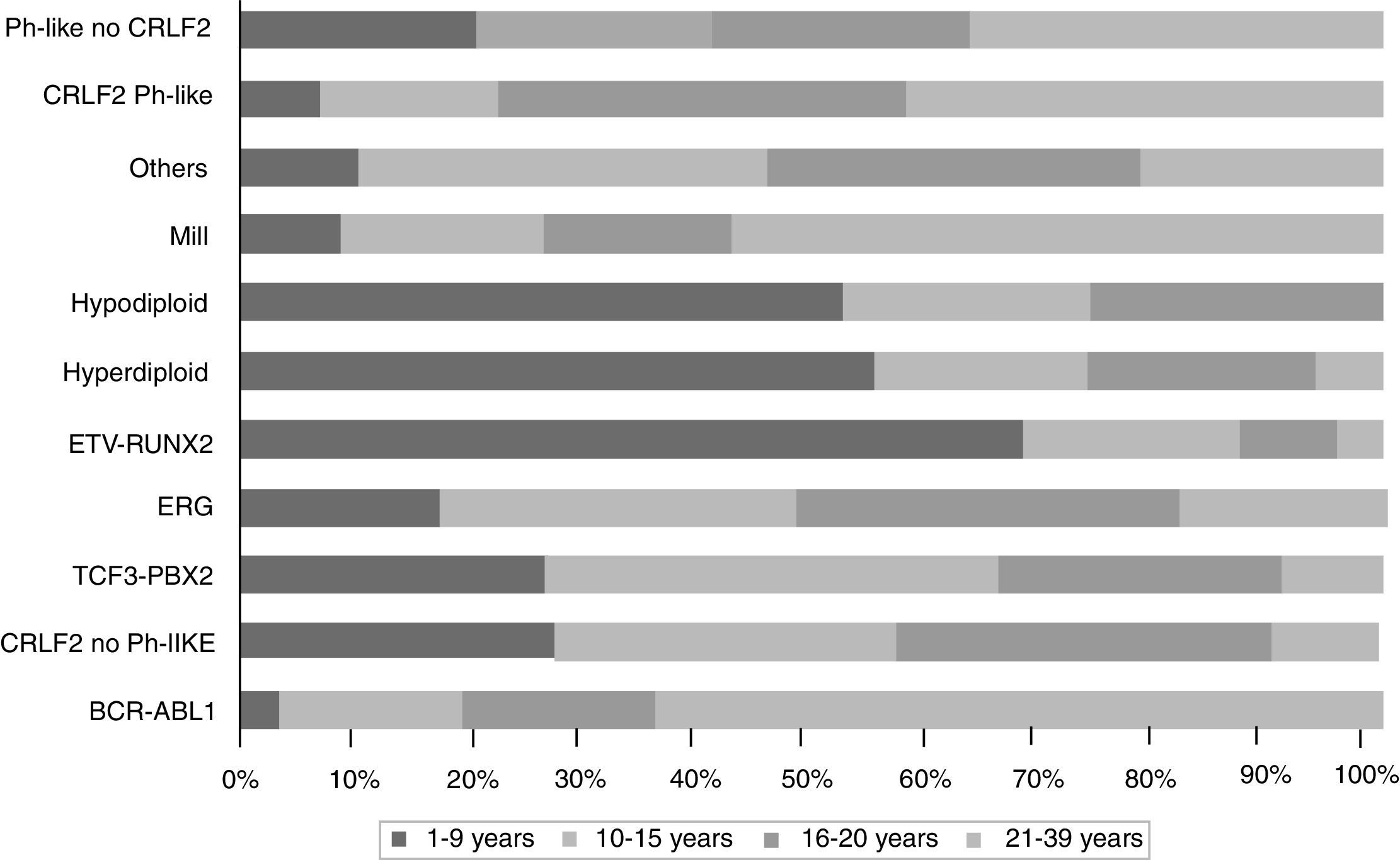
The gene expression in ALL studies, in addition to the analysis of alterations in the number of copies of the DNA, have emphasized the great heterogeneity of the disease and allowed differential diagnosis and identify new subtypes of leukemia.28,33,38,77,78 There is a group of BCR-ABL negative patients (15%) having a gene expression profile similar to that observed in the carriers of this chimeric transcript. This subgroup has a range of genetic and structural alterations of genes that activate lymphoid development, cytokine receptors, and kinase signaling pathways. Over 50% of cases have CRLF1 rearrangements, of which about half present mutations in JAK.38 Recent data show the presence of more than 10 subtypes of pre-B classified as hyperdiploid, hypodiploid, TEL-AML1 positive, TCF3-PBX1 positive, BCR-ABL positive, BCR-ABL like, MLL and MYC rearrangements, with and without rearrangements in CRLF2, deregulation of ERG and alterations in PAX5, which differ in their distribution between age groups (Fig. 1).26,32 Similarly, in the T-cells ALL nine molecular subtypes of this disease, including early pre-T (12%), with deregulation of TLX3 (20%), TAL1 (15-18%), LMO2 (10%), TLX1 (7%), positive for translocations t (10;11) (10%), MLL-ENL (2-3%), NUP214-ABL1 (6%) and t (7;9) (<1%) have been identified.26,32,77 Interestingly, in an analysis of expression profiles in children with early and late relapse it was reported that the overexpression of FOXM1, exonuclease NEF-sp, BIRC5, NCAPH, GTSE1, CENPM, KIAA0101, C10orf56, BUB1B, UBE2V1, POLQ, and TMEM97 are indicators of relapse, while PAICS, TYMS, IMPA2, CAD, ATIC, and GART, correlate with late relapse.26,78
 and its distribution by age (Modified from Mullighan, 2014. Ref. 15).")
Genes that define the different expression profiles in children with ALL are useful in the classification of the disease in adults, particularly for patients with translocations t (1; 19), t (12;21) and 11q23.12,78
7Epigenomics of ALLIn leukemogenesis, epigenetic deregulation plays a key role. Different epigenetic signals for the various subtypes of ALL, in which is very common to find mutations in genes encoding epigenome modifiers, which in turn are associated with therapeutic failure have been described.79–87 Global hypomethylation and hypermethylation of CpG rich islands in the promoter regions of some genes are characteristic of leukemic cells. Hypermethylation and hypomethylation can influence the abnormal expression of genes, promoting or regulating cell proliferation, respectively.86 Genes commonly methylated in ALL include those involved in signaling pathways (TIE1, MOS, CAMLG and GPRC5C), cell cycle regulation and proliferation (cMCTS1 and DGKG), factors and transcription regulators (PROP1, TAF3, H2AFY2, Elf5, ZBTB16, CNOT1 and TADA2A) and homeotic genes (HOXA5 and HOXA6).82
Epigenetic modifications also explain to some extent the resistance to antineoplastic treatments.69 In fact, it has been suggested that the reversible resistance of NOTCH1 inhibitor therapy in T-cell ALL is a phenomenon mediated by epigenetic mechanisms.79 The use of drugs aimed at reversing methylation patterns in specific genes is a potential alternative to anti–leukemic treatment.
Another epigenetic mechanism is the action of non-coding RNAs, which play a pivotal role in cancer biology acting as epigenetic regulators, enhancers and regulating Intra- and inter-chromosomal interactions. Several studies have detected a frequently dysregulated expression of miRNAs: miR -99, -100, 125, -126, -128a / b, -146a, -155, -181a / c, -195, -198, -210, -221, -223, -663 -425-5p, and – 708.81,84–87 Some have been reported as prognostic indicators of treatment response79,80; others have been associated with some leukemic cells lineage, for example, miR-155, -210 and -128b, can discriminate between ALL and acute myeloid leukemia (AML).79,84,85 Overexpression of miR-128b also correlates with ALL with positive MLL-AF4.85 The expression profiles of miRNAs allow to discriminate between lineages81 and can be useful for predicting the response to treatment (miR-10a, -33, -27a, etc.) or monitoring illness after it (Table 3).80–87
Examples of miRNAs with potential clinical significance and its correlation with genomic subtype of ALL.
| Biomarker | ALL type | MicroRNAs | Molecular subtype |
|---|---|---|---|
| Diagnosis | B cells | miR-425-5p, miR-191 and miR-128 | E2A-PBX1 |
| miR-146b and miR-126 | BCR-ABL1 | ||
| miR-383, miR-125b, miR-99a and miR-100 | Hyperploidy and TEL-AML1 | ||
| miR-629 miR-196b, miR-17-92 | MLL-AF4 MLL rearrangements | ||
| T cells Mixed lines | miR-148a, miR-424 and miR-151* miR-196 | ||
| Remission | miR-223 and miR27a | ||
| Relapse | miR-210, miR-708 | ||
| Unfavorable prognosis | miR-33, miR-215, miR-369-5p, miR-496, miR-518d and miR-599 | ||
| Favorable prognosis | miR-10a, miR-134, miR-214, miR-484, miR-572, miR-580, miR-624 and miR-627 |
So far, there is little research that aims to know the interaction between the genome and the environment and its role in the development of ALL. The most explored environmental factors are exposure to X-rays, tobacco, pesticides, insecticides, paint, chlorinated drinking water and alcohol consumption.47,59–61,88–91 The best-studied genes include members of the family of cytochromes, GSTM and MDR. It has been reported that alleles CYP1A1*2A/*2B increase up to five times the risk ALL in children exposed to pesticides during intrauterine development and childhood. Meanwhile, children exposed to high levels of trihalomethanes in drinking water have up to nine times more risk to develop ALL if they are carriers of the CYP2E1 * 5 allele or GSTT1.46 It has also been documented that GSTM1 null genotype and CYP2E1 * 5 allele confers high risk for ALL in children of mothers who consume alcohol during the third trimester of pregnancy and lactation.59 Similarly, the absence of haplotype CGACC (-T1761C, -G9893A, Ex7 + A131G, C1188T, C11599G) of CYP1A1 confers risk for ALL in children who have at least one smoking parent.88 Haplotypes in CYP1A1 and MDR1 (ABCB1) were identified as risk factors for ALL in Hispanic children from the United States exposed to paint and insecticides; CGC haplotype (C1236T, G2677T / A, C3435T) of MDR1 was associated with ALL protection after exposure to indoor insecticides.89,90 Recently, Lupo et al.91 suggested that the rs1804742 of methylene-tetrahydrofolate reductase could confer risk for having children with ALL in mothers with low folic acid intake.
9ALL genomic classification and prognosisFor several years, the clinical, cytomorphological, immunophenotypic and molecular characteristics of leukemia cells have established the diagnosis of ALL and contributed to the design of therapeutic regimens for the disease. According to these features relapse risk is assessed; low-risk patients are treated with less aggressive therapies, while more aggressive and toxic treatments and innovative therapies are reserved for patients with a high risk of relapse.9,32,91,92 The initial mass of leukemic cells, reflected by the LBC, age, and presence of extra–medullary disease are prognostic factors which correlate with the likelihood of disease-free survival (DFS). Children <1 year old, > 10 years of age and those with LBC > 50x109/l have a worse prognosis than younger patients (1-9.99 years) and those presenting LBC <50x109/l. Factors such as infiltration to the nervous system, testicular involvement in males, trisomy 21 or ethnicity can influence prognosis.13,93 Regarding immunophenotype, the ALL pre-B is associated with a more favorable prognosis, whereas co-expression of myeloid antigens in lymphoid blasts (My+ALL) and T cells immunophenotype are factors associated with less DFS.10,39,41
Hyperdiploidy, hypodiploidy, presence of BCR-ABL, TEL-AML1, and alterations involving the MLL gene are the cytogenetic and molecular abnormalities best characterized for prognosis. In vitro studies have shown that hypodiploidy correlates with a DFS of less than 40%, while the hypodiploidy is associated with increased apoptosis and sensitivity to a variety of chemotherapeutic agents; thus, a better response to treatment. The TEL-AML1 expression in some populations is a marker of good prognosis. Carrier cases have a DFS of more than 90% at five years with protocols containing asparaginase. The chimeric transcripts BCR-ABL and MLL-AF4 are associated with a poor prognosis; these patients are candidates for innovative therapies, including medullary transplantation at first remission. In fact, the patients with BCR-ABL are those with greater difficulties because, although a subset of them can be treated with kinase inhibitors (imatinib), the effect is transient and the risk for relapse is very high.10,92,94–96
Great efforts have been made to understand the genetic components of ALL and identify all the genetic lesions that contribute to leukemogenesis and therapeutic failure. Association studies and sequencing transcriptome and genome have shown that patients with pre-B, BCR-ABL1 positive and IKZF1 deletions have a shorter DFS and higher risk for relapse95; cases with deletions in CDKN2A/B without chromosomal rearrangements correspond to a subgroup of patients with a high risk for treatment failure.96 The loss of the tumor suppressor gene CDKN2A/B occurs in over 40% of the cases of ALL pre-B; on the other hand, the SNP rs3824662 of GATA3 has been associated with early response to treatment.95 Other genes linked to relapse are VPREB1, EBF, and ARID5B1.50,78 In this regard, SNPs rs7923074 (OR = 1.2), rs10821938 (OR = 1.2), rs4948488 (OR = 1.41), rs2893881 (OR = 1.45) and rs6479778 (OR = 1.48) in ARID5B are associated with a high risk for relapse in patients of Hispanic and Caucasian origins, being the Hispanic population the one which presents a higher frequency of these variants.50 Furthermore, it has been reported that the presence of deletions and reduced expression of MSH6 correlates with resistance to treatment with mercaptopurine and prednisone and indirectly with relapse.97
Within the group of T-cell ALL, considered with poor prognosis, the patients with alterations that deregulate TLX1 expression (7%) have a low risk of relapse.97
Mei et al.84 suggest that miRNAs also may be useful biomarkers of prognosis and predictors of sensitivity to treatment. For example, the low expression of miR-210 at diagnosis correlates with higher relapse rate or induction failure; when overexpressed in cell lines REH and RS4;11, these are more sensitive to treatment with daunorubicin, L-asparaginase, and vincristine.
10Therapeutic targetsIdentifying chromosomal abnormalities in hematologic malignancies has been fundamental in the discovery of therapeutic targets for tackling the disease. One of the first successful treatments for leukemia was the use of retinoic acid in patients with acute promyelocytic leukemia carriers of t (15:17) (q22;q12) (PML-RARA).98 Subsequently, the identification of a tyrosine kinase inhibitor (imatinib) allowed efficiently to treat patients positive for t (9;22) (q34,q11) (BCR-ABL) with chronic myelocytic leukemia and ALL.99
Some other treatments are in clinical phase 1; for example, the inhibitors of JAK (ruxolitinib) in patients with CRLF2 rearrangements and mutations in JAK1 and JAK2.
Identifying changes in expression of genes also offers opportunities for the development of new therapeutic targets. For example, the subtype of T cells ALL, which has an expression profile similar to that of normal thymocytes in early stages of differentiation, have a very poor prognosis. Therefore, it is suggested that these cases should be treated with high–doses of dexamethasone.99 Recently, it was reported that TPMT genes (mercaptopurine, thioguanine, azathioprine), CYP2D6 (Codeine, tramadol, oxycodone, amitriptyline, ondansetron, fluoxetine, paroxetine), CYP2C19 (clopidogrel) and SLCO1B1 (simvastatin potentially methotrexate) have been included in the electronic medical record for clinical implementation, being the of TPMT genotype the one that has a greater relevance to the treatment of children with ALL.100–104
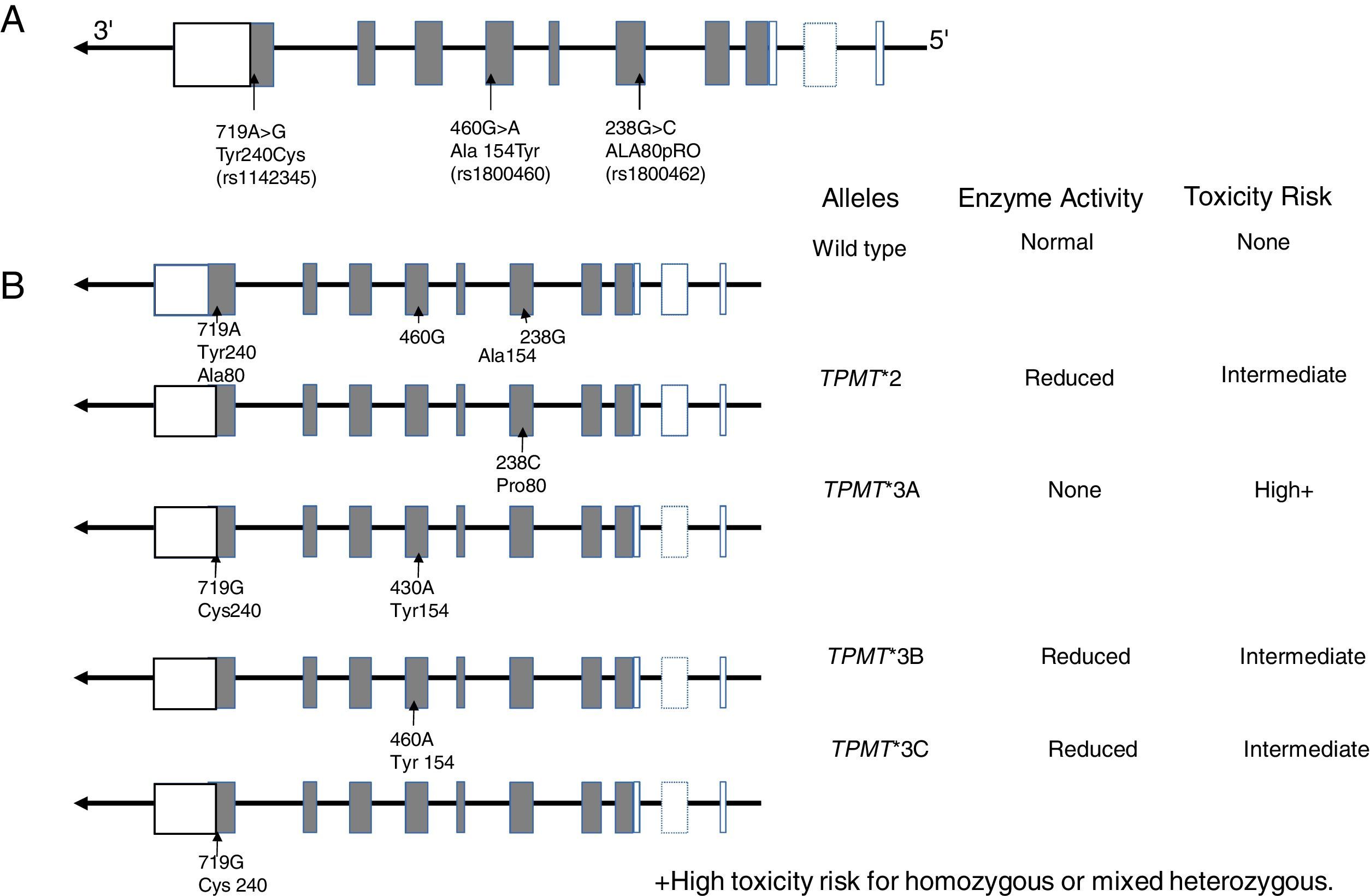
10.1TPMT geneTPMT is an enzyme localized in the cytoplasm involved in the metabolism of purine analogs used in anti-leukemic and immunosuppressant treatments such as 6-mercaptopurine (6-MP), thioguanine (6-TG) and azathioprine. TPMT inactivates thiopurines, thus in its absence, levels of thioguanine nucleotides (TGNs) active metabolites increase in hematopoietic cells, thereby conferring a high risk of hematotoxicity that endangers patient's life. It is now known that TPMT variants rs180462 (c.238G> A), rs1800460 (c .460G> A) and rs1142345 (c.719A>G) correlate with this enzyme's activity and are predictors of toxicity and effectiveness of treatments based on thiopurines, having substantial clinical significance in the treatment of patients with ALL and with other autoimmune entities. Alleles TMPT * 2 (c.238G> A, Arg80Pro) cause a reduction in this enzyme's activity; patients who are heterozygous for a mutant allele have intermediate risk of toxicity, while homozygous and compound heterozygous show very low or undetectable enzyme activity, and the risk of a toxic effect is very high (Fig. 2). These patients should be treated with lower doses of mercaptopurines to decrease toxicity and not to compromise the treatment; a reduction of 90% in the dose for homozygous patients and of 30% to 70% in carriers of a single deficient allele is suggested.101–103
11Treatments in preclinical phases The structure of the gene and location of the polymorphisms. Boxes represent exons of the noncoding region (white) and coding region (gray); B) Alleles affecting enzyme activity.")
Despite the major advances in the treatment of ALL, a percentage of cases die as a result of the illness or treatment-associated side effects. Identifying new therapeutic targets may allow the development of new alternative therapies: inhibition/induction of altered genes, immunotherapy, among others.105–108In ALL patients showing an expression profile similar to BCR-ABL and which carry gene fusions EBF1-PDGFB or NUP214-ABL1, imatinib, an inhibitor of ABL and PDGFB, is in preclinical evaluation.105
T cells ALL is a rare and extremely aggressive entity; carriers of mutations in the gene NOTCH1 do not respond to conventional treatments and have a high rate of relapse and mortality. For their treatment, NOTCH1 inhibitors are in development (GSIs), which have not been brought into clinical phase because there is experimental evidence that the leukemic cells are resistant to treatment with GSIs, a mechanism probably mediated by epigenetic phenomena.106
Other target genes for the treatment of the T cells ALL are BDR4 and BCL2. BDR4 is a protein found over expressed in this neoplasia and described as a potential promoter for the expression MYC and the anti-apoptotic molecule Bcl-2. In fact, as an alternative treatment for T cells ALL, the combined use of inhibitors of NOTCH1, BDR4 and BCL2 has been proposed.107
Immunotherapy as an alternative treatment is of recent interest; the therapeutic targets are the surface antigens of leukemic blasts. For this purpose, naked and unconjugated antibodies and immunotoxins are being developed, as well as receptors for chimeric antigens and single chain bispecific antibodies that couple to T lymphocytes.108
12Genomics and ALL in MexicoThe application of genomic technologies in the childhood ALL research is adding a plethora of data confirming their genomic complexity. Also, they are contributing to the detection of biomarkers that are potentially useful for detecting patients at high risk for relapse or treatment toxicity before clinical manifestations of the disease are present. Currently, NGS platforms allow the study of groups of genes that are commonly affected by cancer who are carriers of drivers mutations, for which there are pharmacological treatments in preclinical stages (inhibitors of JAK in patients with rearrangements in CRLF2 and mutations in JAK1 and JAK2). The affected genes can be used as biomarkers for MRD.106,107
In Mexico, ALL genomic research suggest FLT3, DEFA1, and hsa-miRNA-511 as potential markers of risk for relapse or response to treatment33; however, this data should be replicated in independent populations before its implementation in the clinical practice. It has been widely demonstrated that the detection of BCR-ABL, and TPMT genotyping should be performed in all children with ALL at diagnosis, and that the first treatment option for carriers of BCR-ABL is imatinib mesylate instead of bone marrow transplantation.4,6,102 In the case of carriers of one (heterozygous) or two (homozygous or compound heterozygous) TPMT null alleles, which together are about 8% of Mexican children with ALL,109,110 the dose of thiopurines should be decreased 30 to 90% to reduce the risk of myelosuppression and the potential death of the patients.99–104 However, although there have been great efforts, the use of both biomarkers in the clinical practice is still insufficient in our country. We are not far from constituting them as diagnostic tools in the clinical management of patients treated in public hospitals, but for now, the molecular study, particularly for the detection of BCR-ABL at diagnosis, is only available in some third level hospitals.16–18 The challenge is that physicians and researchers add efforts to find routes that facilitate the optimization of technical resources and infrastructure to enhance the translation of genomic research to personalized medicine in Mexican children with ALL.
“Omic” tools have revealed that, in addition to somatic alterations acquired recognized as markers of ALL (aneuploidies, translocations as BCR-ABL, TEL-AML1), germline or inherited genetic variations have important implications for ALL risk in drug response or the toxicity of antineoplastic therapies. There are clear examples such as the use of TPMT genotype in the treatment of children ALL, but more studies that integrate genetic, somatic and germline information to predict the risk of relapse and detecting MRD are needed, with the ultimate goal of applying this knowledge and provide timely treatment and personalized medicine. Thus, subsequent genomic studies should incorporate clinical, pathological, and molecular information to identify alleles that contribute to the malignancy of human entities.
FundingNone
Conflict of interestThe authors declare no conflicts of interest of any nature.
Please cite this article as: Jiménez-Morales S, Hidalgo-Miranda A, Ramírez-Bello J. Leucemia linfoblástica aguda infantil: una aproximación genómica. Bol Med Hosp Infant Mex. 2017;74:13–26.