





Alpha tricalcium phosphate (α-TCP) is a ceramic widely used in cements for orthopedic and dentistry applications, which is synthesized by solid-state reactions occurred at high temperature, being highly relevant the control of the cooling rate in order to avoid the formation of secondary phases as beta tricalcium phosphate (β-TCP). From the above, the effect of TCP cooled at three different rates on the properties of the derived calcium phosphate cements was studied. The results show that a low cooling rate of 10°C/min is enough to avoid the formation of the beta phase. Additionally, remarkable differences among the final calcium phosphate cements derived from the differently cooled TCP ceramics in terms of compressive mechanical resistance, final phases and chemical bonds, were not evidenced.
El fosfato tricálcico alfa (α-TCP) es un cerámico ampliamente utilizado en cementos para aplicaciones ortopédicas y odontológicas, el cual es sintetizado por reacciones de estado sólido que ocurren a alta temperatura, donde es de alta importancia el control de la velocidad de enfriamiento para evitar la formación de fases secundarias como el fosfato tricálcico beta (β-TCP). De lo anterior, fue estudiado el efecto del TCP enfriado a tres velocidades distintas sobre las propiedades de los cementos de fosfato de calcio derivados. Los resultados muestran que una velocidad de enfriamiento de 10°C/min es suficiente para evitar la formación de la fase beta. Adicionalmente, no se evidenciaron notorias diferencias entre los cementos de fosfato de calcio finales derivados de cada TCP enfriado diferentemente en términos de resistencia mecánica a la compresión, fases finales y enlaces químicos.
Tricalcium phosphate (TCP) is of great interest among calcium phosphates (CaPs) as it can be used directly as a bone replacement biomaterial, as a promoter of calcium phosphate cements (CPC) or combined with other CaPs for biomedical applications [1,2]. TCP presents three polymorphs with specific phase transformation temperatures. According to Kreidler and Hummel's phase diagram [3], β-TCP (rhombohedral) transforms at 1125°C toward α-TCP (monoclinic) and at 1430°C α-TCP converts toward α′-TCP (hexagonal). The α′-TCP is only stable above such high temperature and reversion toward α-TCP occurs immediately when cooling from it. Instead α-TCP is metastable at room temperature and discussion about the cooling rate required to avoid reconversion to β-TCP is wide, several researchers indicate the need of rapid quenching to achieve it [4–8], while others indicate that using low cooling rates as 5°C/min is enough to avoid such phase transformation [9–12]. Discussion also relies on the content of impurities that can stabilize either the α polymorph e.g., silicon (Si) or others the β polymorph e.g., magnesium (Mg) and strontium (Sr) [10,12–15].
Despite these researches have analyzed the effect of cooling rates on TCP, none analysis has been reported regarding the influence of the cooling method to obtain TCP derived CPCs. CPC may be produced by one or a combination of several CaPs but mainly the base component used in CPCs is α-TCP and, as shown by several researchers, the particle size and the liquid to powder (L/P) ratio have an effect on the final morphology of calcium deficient hydroxyapatite (CDHA) and the porosity of CPC after setting [16–19]. Reports show that when particle size increases porosity and specific surface area decreases and when the L/P ratio increases porosity augments and mechanical properties decreases. Nevertheless, the effect that may be caused on CPC physicochemical properties, mechanical behavior and microstructural homogeneity by the TCPs, cooled by different methods during thermal synthesis, have been not assessed in those researches.
In this work, the focus has been on analyzing the effect of the cooling rate on the TCP obtained in terms of chemical and phase characteristics as well in each TCP derived CPC. Also, a thermal analysis on the starting reagents for the TCP thermal synthesis was assessed to understand the thermal events during heating and cooling of the material. Differences in the TCPs and derived CPCs obtained are compared to evaluate if a low cooling rate inside the furnace is optimal to produce a α-TCP with a calcium to phosphorous (Ca/P) ratio around 1.5 and subsequently a CPC with CDHA crystals and a mechanical resistance close to the one of trabecular bone (5–45MPa [20]).
Materials and methodsThe reagents used to prepare the TCPs were characterized, then three TCPs were obtained through different cooling methods and characterized for further CPCs preparation and final characterization.
Raw material characterizationChemical analysis of the starting reagents was carried out to identify possible impurities that may affect the stability of the α and β-TCP phases at high temperature using the wavelength dispersive X-ray fluorescence (WDXRF, ARL Optim′X ThermoFisher, USA).
In addition, to understand the thermal phenomena occurring in the reagents mixture of calcium hydrogen phosphate (CaHPO4, Sigma-Aldrich) with calcium carbonate (CaCO3, Merck) at a molar ratio of 2:1 during heating and cooling inside the furnace, a thermal gravimetric analysis coupled to differential scanning calorimetry (TG-DSC, NETZSCH STA 449F3, Germany) was performed. Mass loss and thermal exchanges were identified during heating and cooling at 10°C/min in the range of room temperature to 1500°C, using air at a flow rate of 20mL/min to mimic furnace conditions; 60.92mg of the mixture were analyzed on a platinum–rhodium crucible.
TCPTricalcium phosphate was obtained through high temperature solid-state reaction. It consisted of first mixing CaHPO4 with CaCO3 at a molar ratio of 2:1 inside a plastic container and then homogenizing was performed by placing the container inside a Ro-Tap (vertical sieve shaker) at 750rpm. Subsequently the mixture was poured in a platinum crucible, placed inside a furnace and heated until 1400°C (sustainment time of 2h) and finally cooling process was performed in accordance with the desired cooling method. Three cooling methods were performed with the purpose of comparing the effect of the cooling rate on the TCP obtained. The slowest method consists in cooling the material inside the furnace at a rate of 10°C/min (TCP-F) as tested in some previous researches [9,21,22], the intermediate method consisted in performing a quenching of the material using an air jet (TCP-A) [23,24] and the fastest method is achieved by applying air jet assisted by striking the material (TCP-S) with a stainless steel hammer against a stainless steel base. The strikes break the material, which is partially sintered, and increase the TCP cooling surface so making the cooling happens faster. Each TCP was milled in a 250mL zirconia ball mill (S100 Centrifugal Ball Mill, Retsch GmbH, Germany) using zirconia balls (four of them 20mm in diameter, five of 15mm and five of 10mm) for 10min at 350rpm. Finally the TCP powder was sieved trough a 25μm hole size mesh (#500 mesh, Pinzuar LTDA, Colombia).
Laser scattering particle size analysis (Mastersizer 2000E with Hydro 2000 MU accessory, Melvern, USA) using analytic grade ethanol (Sigma-Adlrich) was used to assess and compare the particle size distribution of the three TCP samples synthesized. In order to identify the possible differences in the elemental composition and the polymorphs in the three obtained TCP, analysis by wavelength dispersive X-ray fluorescence (WD-XRF, ARL Optim′X ThermoFisher, USA) as well as by Fourier Transformed Infrared Spectrometry (FTIR-DRIFT, IRTracer, Shimadzu, Japan) were carried out. X-ray diffraction was performed (XRD; Empyrean, PANanalytical B.V., The Netherlands) with a copper radiation source (Cu Kα, λ=1.540598Å), operated at 45kV and 40mA and analyzed in the 2θ range of 20–40° at a scan step size of 0.02° and a time per step of 60s in order to detect the crystalline phases. Phase identification was performed with software Xpert Highscore 2.0 (Empyrean, PANanalytical B.V., The Netherlands) using the reference patterns of the Joint Committee on Powder Diffraction Standards (JCPDS) 9-348, 9-169 for α-TCP and β-TCP respectively.
CPCFrom each TCP powder obtained a CPC was prepared by adding the specific volume of deionized water to obtain a L/P ratio of 0.44mL/g, followed by vigorous agitation during 1min and posterior pouring into Teflon cylindrical molds (6.0mm in diameter and 12.0mm in height) for compression test in accordance to ASTM C1424-15. Samples were allowed to set immersed in Ringer's solution during 7 days at 37°C and after this time dried at 60°C for 24h and stored inside desiccator until the mechanical testing. CPCs are labeled as CPC-S, CPC-A and CPC-F in accordance with the cooling method (with air jet complemented with strikes, with an air jet and inside a furnace respectively) used to obtain the TCP samples.
Compression tests were performed in a universal testing machine (Shimadzu AGS-X 50kN, Japan) at a crosshead speed of 0.5mm/min. After compression tests, the fracture surface of CPCs was analyzed using a scanning electron microscope (JSM-6490LV, JEOL Ltd, Japan) to compare the morphology, the porosity and the homogeneity of the calcium deficient hydroxyapatite (CDHA) crystals in the obtained cements. Samples after compression tests were crush on an agata mortar for FTIR and XRD analysis (performed as previously described in the TCP characterization). Phase identification was performed using the software Highscore Plus 3.0 (PANalytical B.V., The Netherlands) using the reference patterns of the Joint Committee on Powder Diffraction Standards (JCPDS) 9-348, 9-169 and 9-432 for the α-TCP, β-TCP and CDHA respectively. Finally, phase quantification of the cements was performed by Rietveld refinement with the materials analysis using diffraction free-software (MAUD, University of Trento, Italy); results were confirmed by a residual in weight (Rwp) lower than 10% and a significance level equal or lower than 0.02 [25].
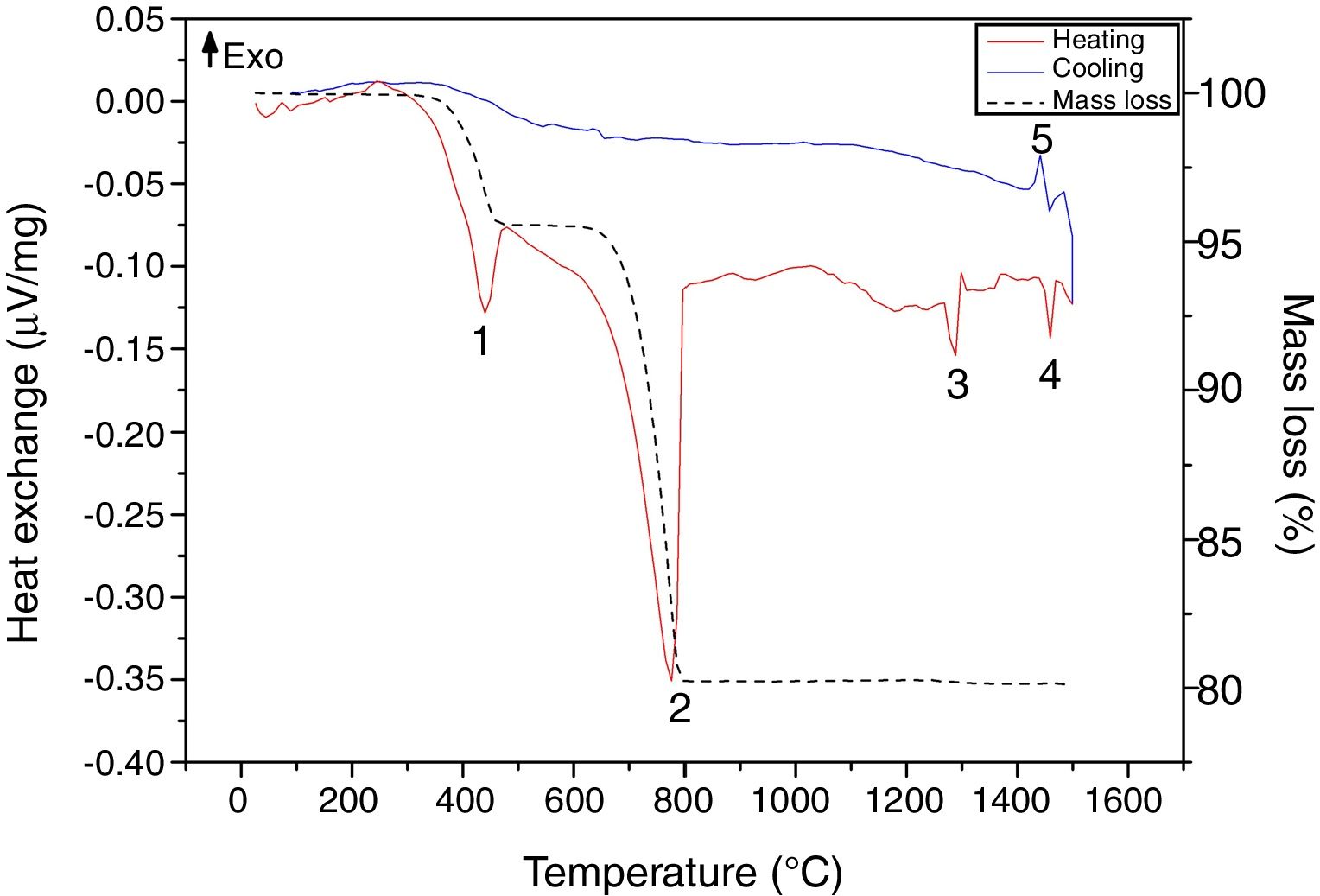
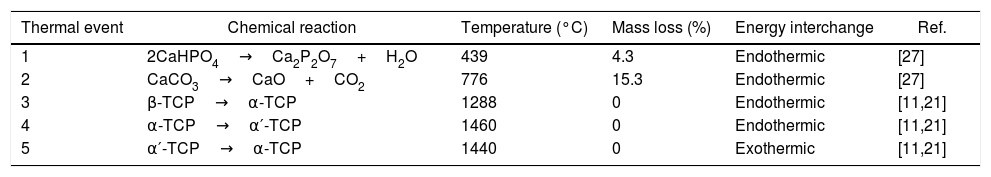
Results and discussionRaw material characterizationTGAFig. 1 shows the TG-DSC of the starting reagents at the specific molar ratio of 2:1. Four endothermal events can be identified during heating. The two first are associated to the reagents decomposition, first CaHPO4 produces calcium pyrophosphate (Ca2P2O7) at 439°C and second CaCO3 produces CaO at 776°C. These two products will react in the region between 780 and 1100°C to produce β-TCP as follows: CaO+Ca2P2O7→β-Ca3(PO4)2, however such event is not identified in the DSC, since it is a process with low interchange of energy and is under the sensitivity of the technique and then, thermal mechanical analysis (TMA) is recommended instead [26]. The third event occurs at 1288°C and is attributed to the β→α phase transformation, crystalline network changes from rhombohedral to monoclinic. Then, the fourth event at 1460°C where α→α′ is presented, here a hexagonal cell distribution appears. However, during the cooling process only one exothermic peak is visible, which is related to the reconversion of α′→α at 1440°C. Both heating and cooling processes were performed at 10°C/min and no reconversion of α→β is identified, it can be confirmed that cooling at such rate inside a furnace with an air atmosphere will not lead TCP to the non-desired β phase. The total mass loss of 19.8% is associated to water evaporation (0.2wt.%) and to CaHPO4 and CaCO3 decompositions (4.3 and 15.3wt.% respectively). Further phase transformation events are not related with mass loss as only crystalline network is changing. The thermal processing of the powder mixture promotes a minor sintering of particles but no melting occurs in the analyzed temperature range. Table 1 summarizes all identified thermal events in the TG-DSC assay.
. DSC during heating (red line) and cooling (blue line). Mass loss (black dash line).")
Some researchers have demonstrated that impurities in the starting reagents as magnesium (Mg) and strontium (Sr) are β stabilizers and silicon (Si) is α stabilizer at high temperatures processing. XRF shown in Table 2 indicates that the chemical composition of the reagents was mainly P2O5 and CaO. The low contents of MgO (0.13wt.%), SrO (0.02wt.%) and SiO2 (0.01wt.%) cannot be considered as phase stabilizers impurities as values are below the ones established by others researchers [13,15,23,28,29]. TG-DSC and chemical content corroborate that quenching is not needed when α-TCP is the only phase desired via thermal processing. As stated by Monma and Goto [30], high energy is required to promote the β→α phase transformation and such energy is also required for reconversion, meaning that the cooling rate of 10°C/min is fast enough to avoid high content of β-TCP in the final desired phosphate.
Chemical composition (wt.%) of starting reagents for solid state synthesis of TCP.
| Reagent | P2O5 | CaO | Na2O | MgO | Al2O3 | K2O | WO3 | SrO | SiO | S | NiO | Cr2O3 | Fe2O3 | LOI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CaHPO4 | 49.5 | 43.0 | 0.18 | 0.13 | 0.12 | 0.02 | 0.01 | 0.02 | 0.01 | N.D. | 0.01 | N.D. | 0.02 | 7.0 |
| CaCO3 | N.D. | 59.0 | N.D. | N.D. | N.D. | N.D. | 0.02 | 0.01 | N.D. | 0.01 | 0.01 | N.D. | N.D. | 41.0 |
N.D.: non detected.
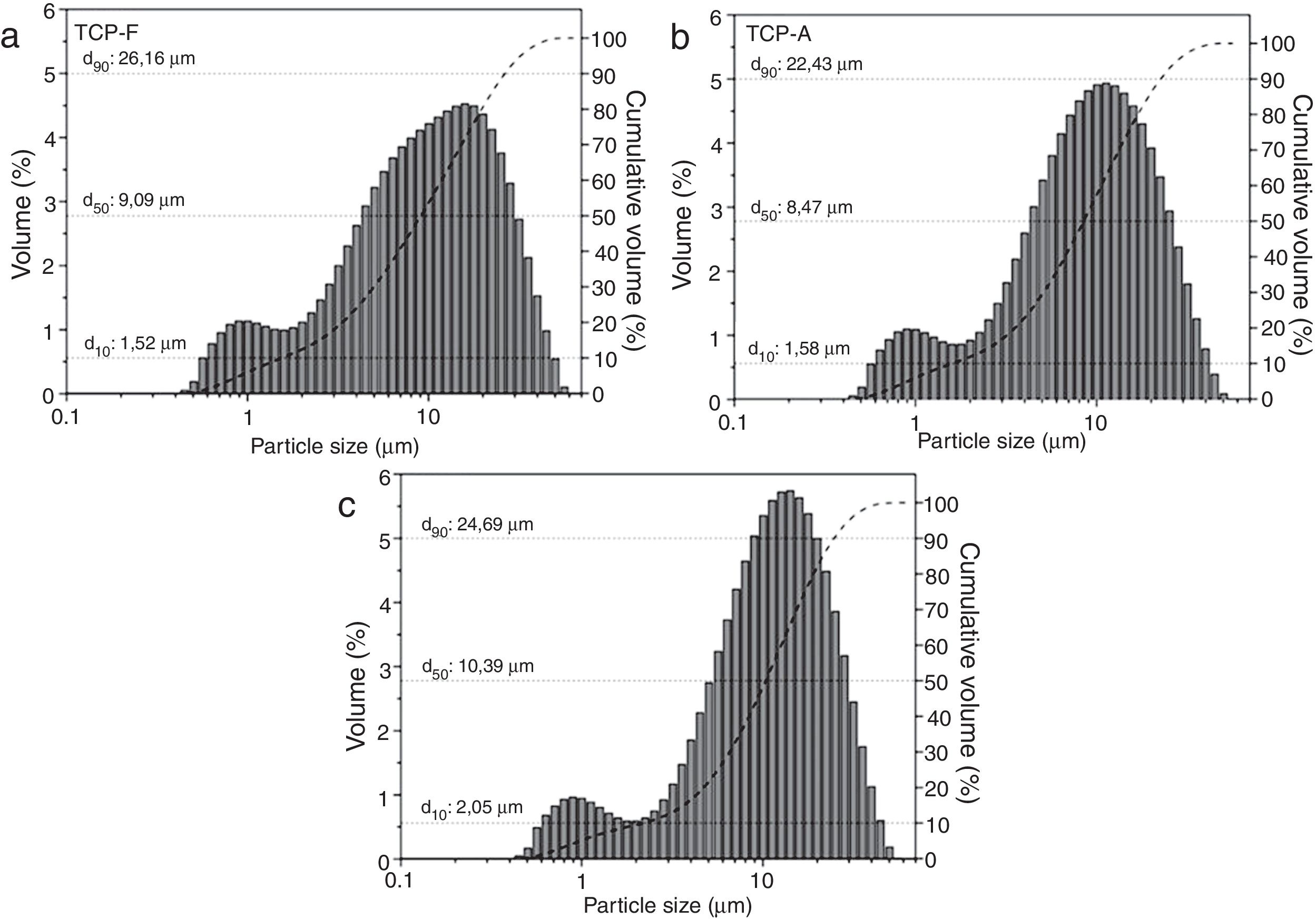
Particle size distribution (PSD) of the three TCP samples is shown in Fig. 2. It is clear that all TCPs present a bimodal and similar distribution. In Fig. 2a, TCP-F shows a PSD of d10: 1.5μm, d50: 9.1μm, d90: 26.2μm; while the PSD for TCP-A shows in Fig. 2b present a slightly lower amount of big particles being d10: 1.6μm, d50: 8.5μm, d90: 22.4μm and Fig. 2c shows the PSD for TCP-S, which present d10: 2.0μm, d50: 10.4μm and d90: 24.7μm. These similar PSD indicate that the milling and sieving process affected equally all TCPs. As shown in several works, particle size distribution affects reactivity of TCP and further the microstructure of the derived CPC [16–18,31–33], such d50 ranging 8.5 and 10.4μm will lead to plate like CDHA crystals [34], and further no reactivity differences will occur due to the similar particle size distribution.
XRF of (a) TCP-F, (b) TCP-A and (c) TCP-S, cumulative volume percentage (dash line) and its respective d10, d50 and d90 identification.")
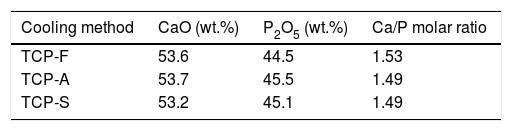
Elemental composition and Ca/P ratio of the three TCPs samples is presented in Table 3, indicating similar Ca/P ratio around 1.5, which is a characteristic value of pure TCP and is the appropriate one to promote CDHA (Ca/P=1.5) precipitation after conversion of TCP [35]. All TCPs present mainly CaO and P2O5 with a sum of weight percentage (wt.%) higher than 98%. The remaining oxides are as follows, TCP-F (SiO2: 0.58; Al2O3: 0.21; Fe2O3: 0.15; MgO: 0.1, ZrO2: 0.16wt.%, other oxides below 0.1wt.%), TCP-A (Al2O3: 0.44; Na2O: 0.15wt.%, other oxides below 0.1wt.%) and TCP-F (SiO2: 0.15; Al2O3: 0.29; Na2O: 0.13; ZrO2: 0.3wt.%, other oxides below 0.1wt.%).
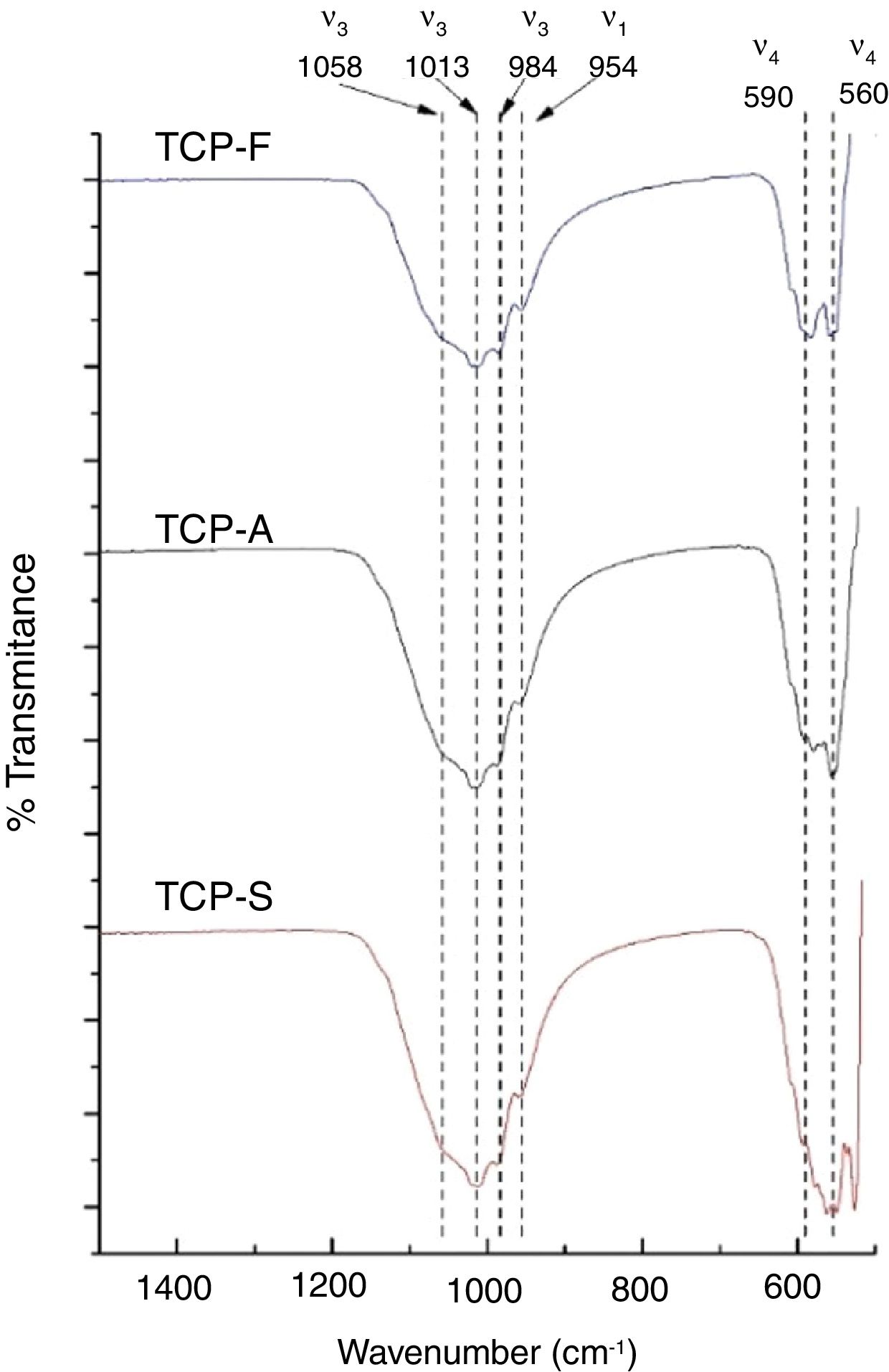
FTIRFTIR-DRIFT TCPs spectra shown in Fig. 3 present the characteristics bands of the PO4− ion. The most intense bands are obtained at 1058, 1013 and 984cm−1 for the antisymmetric PO stretching bond (ν3), at 954cm−1 for the symmetric PO stretching bond (ν1) and a doublet at 560–590cm−1 for the antisymmetric PO bending bond (ν4) [11]. The main difference is seen in the low wavenumber doublet, where greater distortion is observed as the cooling method increases. The distortion of the phosphate ion is attributed to a lower order of crystallinity in TCPs [36,37], which is in consistent as a faster cooling method will lead to a less crystalline material.
XRD
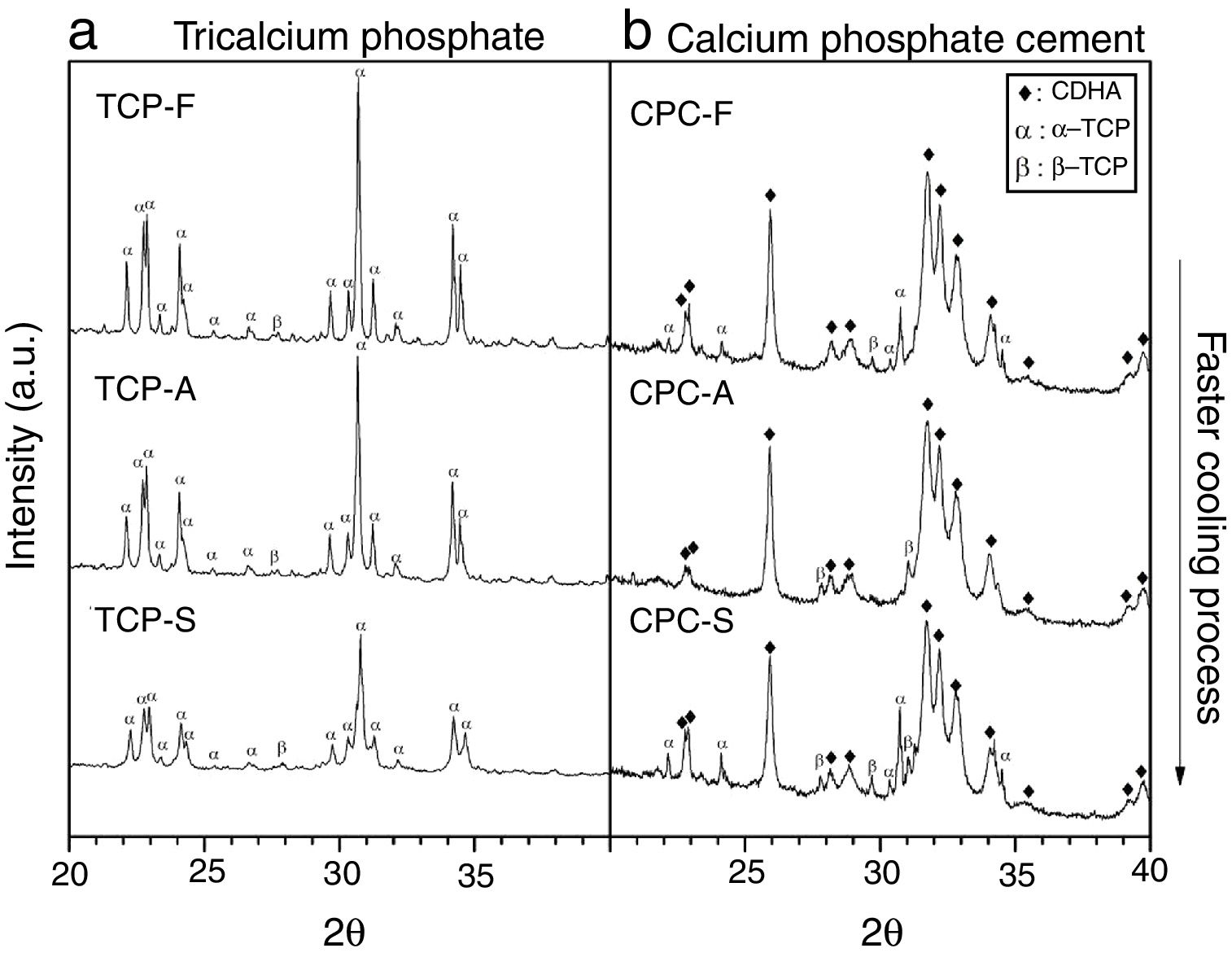
Fig. 4a shows the X-ray diffractogram of the TCPs samples obtained by the three cooling methods, where identified peaks are mainly related to α-TCP and only one peak is related to β-TCP (2θ: 27.8°) in accordance with patterns JCPD 9-348 and JCPD 9-169 respectively. This result indicates that the alpha phase is highly obtained in the three TCPs and that even by cooling the TCP at a low rate of 10°C/min will lead to the desired CPC precursor. This result is in accordance with those described by others authors [9,12,13,21], which indicate that in specific conditions where beta stabilizers as magnesium or strontium oxides are not present in the reagents a low cooling rate is enough to avoid the phase transformation from the alpha to the beta TCP. However, the only identified difference among the TCPs is a broadening in the base of the peaks as the cooling method is faster. Hence, the TCP cooled inside the furnace presents the sharpest peaks; such phenomenon is attributed to a difference in crystallinity, and agrees with the result obtained by FTIR, confirming that the faster the cooling method, the less crystalline the TCP is.
CPC characterizationXRD the three TCPs samples (left column) and b) its derived CPCs (right column). Phases identified by patterns JCPD 9-432 (CDHA), 9-348 (α-TCP) and 9-169 (β-TCP).")
Fig. 4b shows XRD spectra of the TCP derived CPCs after 7 days of setting at 37°C in Ringer's solution. It has been identified that the final CPCs present peaks associated to CDHA, some to α-TCP and to traces of β-TCP by comparison with XRD patterns JCPD 9-432, 9-348 and 9-169 respectively. As the setting process starts with α-TCP dissolution and followed by CDHA precipitation, it is clear that α-TCP conversion in CPC-F and CPC-S is not culminated after 7 days of setting as given that principal peak at 31.7° (2θ) is still expressed. In CPC-A it is notorious that almost full conversion was obtained. As some β-TCP was detected in all TCPs previous to the cements preparation (Fig. 4a), such phase outstands more in the CPCs as its solubility is much lower than α-TCP's one.
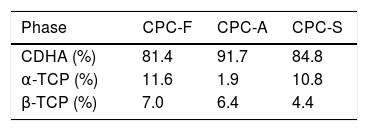
Table 4 shows the phases quantification of each CPC sample. CPC-F and CPC-S have the lowest amounts of CDHA (81.4 and 84.8wt.% respectively), whilst CPC-A has the highest (91.7wt.%). This indicates that TCP-A medium crystallinity in comparison with the other two TCPs is the most appropriated to favor the α-TCP dissolution and the subsequent CDHA precipitation. However, it was expected that less crystalline TCP showed higher reactivity of α-TCP and a higher amount of CDHA in the final CPC, as some works state that amorphous CaPs are highly reactive [37,38].
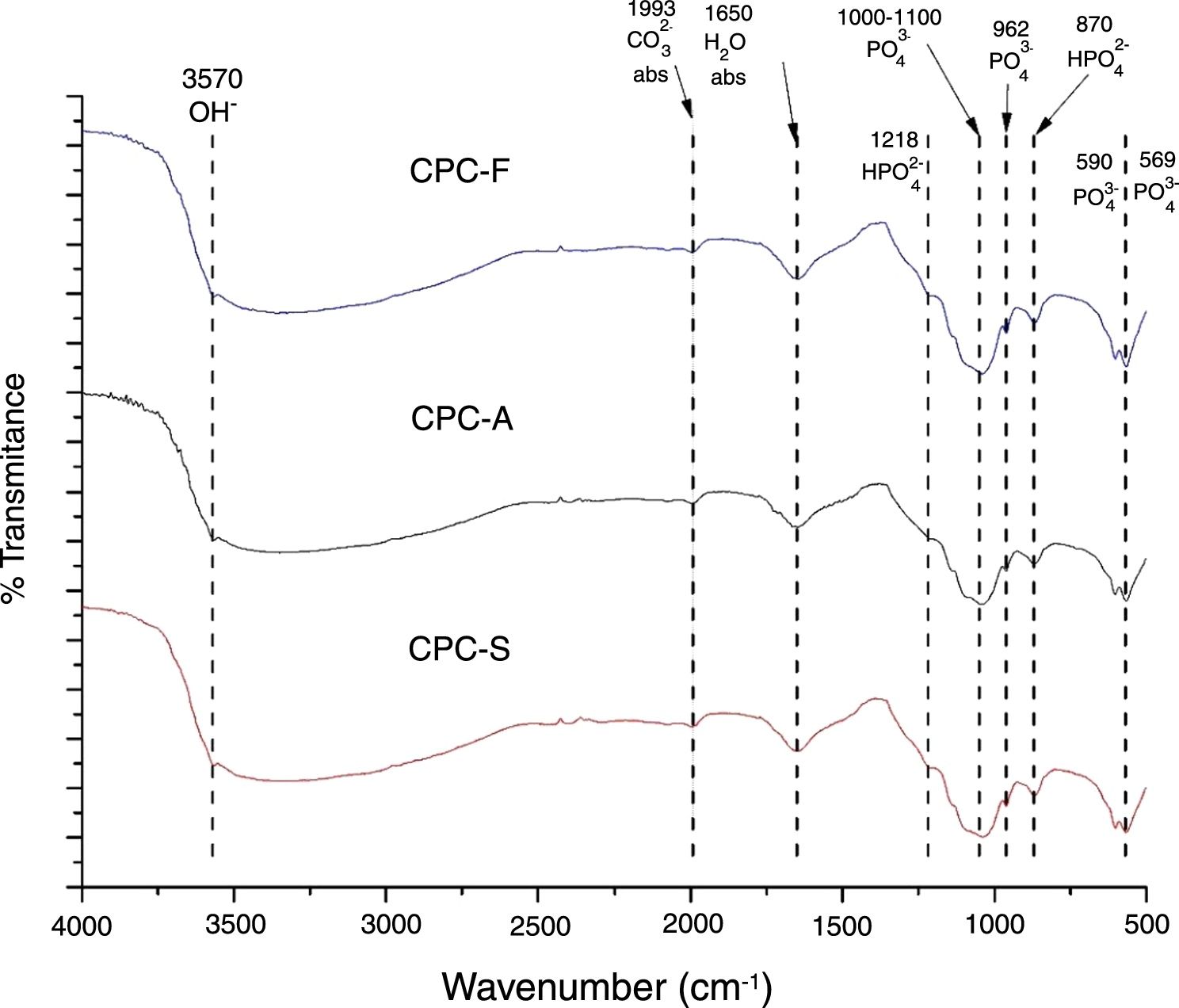
FTIRThe FTIR spectra shown in Fig. 5 exhibits the three samples TCP derived CPC. It is clear that no differences among the CPCs is presented, indicating that the CPCs are similar in terms of chemical bonds; PO43− is identified by 569, 590, 962cm−1 bands and a broad band between 1000 and 1100cm−1. HPO42− is identified by bands at 870 and 1218cm−1. H2O and CO32− absorption are also identified by 1650 and 1993cm−1 bands respectively. OH− band at 3570cm−1 indicates that formation of hydroxyapatite is present in the CPCs. Presence of HPO42− bands and the absorption of H2O and CO32− on CPCs is common when the setting is performed by immersion on Ringer's solution, indicating that the hydroxyapatite obtained is a calcium deficient type, CDHA [39,40].

The differences between the FTIR spectra of TCPs (Fig. 3) with its respective CPCs (Fig. 5) show that conversion of the starting phosphate is occurring in favorable way to promote CDHA precipitation.
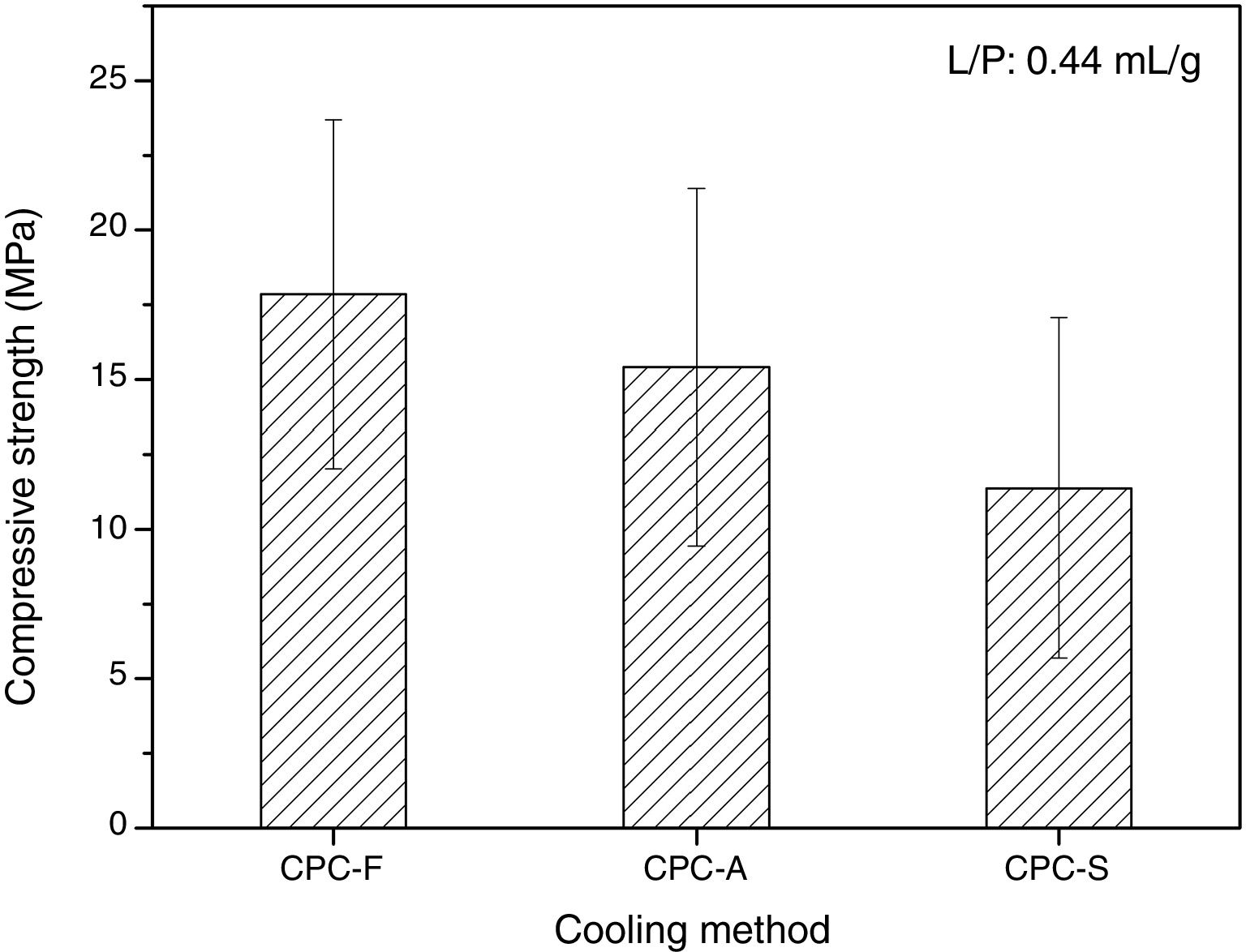
SEM and compression testsIn Fig. 6, the SEM images show the morphology of the three CPCs samples in their fracture surfaces produced by compression tests, where at low magnification (500×) CPC-F and CPC-S have similar topography and cements seem highly dense. However, in CPC-A it is clear that higher porosity was obtained, which may explain why CDHA content was higher, since water could enter more easily inside the sample and promote higher dissolution of the starting α-TCP. At higher magnification (10,000×) characteristic CDHA fine plate like crystals may be observed in all cements. More ordered crystals are present in CPC-F and more disordered on CPC-A and CPC-F, perhaps due to the higher reactivity of their TCPs precursors. Despite these low morphological differences, CPCs maximum compressive strengths (Fig. 7) evidence no statistical difference among them three. However, average values seem to decrease as CPC is derived from faster cooled TCP; CPC-F (18±6MPa), CPC-A (15±6MPa) and CPC-S (11±6MPa), which would be explained by the higher ordered CDHA that implies greater crystals entanglement and then, higher mechanical resistance of the CPC [41–44].
, inserted images at 10,000× (scale bar 1μm).")

TCP may be obtained by solid-state synthesis of 2CaHPO4:CaCO3 mixture and cooled by three different methods. Phases analysis showed that α-TCP was the main identified phase, indicating that even at low cooling rates from 1400°C like 10°C/min, the α phase will prevail and no reconversion to the β polymorph will occur, especially when impurity amounts in the starting reagents are below the needed for beta-stabilization (1.65% MgO and 1.0mol% SrO). TG-DSC result demonstrated that the α to α′ phase transformation is a enantiotropic phase transition as peaks during heating and cooling around 1460°C were identified. Meanwhile β to α transformation is only monotropic as no peak during cooling was identified close to 1200°C. However, TCP crystallinity seemed to decrease as the cooling method was faster, as indicated by the broadening of XRD base peaks. Differences in the derived CPCs were only related to higher CDHA content and porosity in CPC-A. Also, more ordered CDHA crystals appear in the CPC as the starting TCP is more crystalline due to a lower dissolution rate of the phosphate, nevertheless, the compression resistance of the three CPCs is not different. This work shows that a non-quenched TCP obtained by cooling it at 10°C/min will lead to a CPC with similar physicochemical and mechanical characteristics as a CPC derived from a TCP obtained by quenching processes.
The authors acknowledge the Colombian Administrative Department of Science, Technology and Innovation (COLCIENCIAS) for the fundings given by the project 1115745-57862.

