


Human activities on the Earth's surface change the landscape of natural ecosystems. Mining practices are one of the most severe human activities, drastically altering the chemical, physical and biological properties of the soil environment. Bacterial communities in soil play an important role in the maintenance of ecological relationships. This work shows bacterial diversity, metabolic repertoire and physiological behavior in five ecosystems samples with different levels of impact. These ecosystems belong to a historical area in Iron Quadrangle, Minas Gerais, Brazil, which suffered mining activities until its total depletion without recovery since today. The results revealed Proteobacteria as the most predominant phylum followed by Acidobacteria, Verrucomicrobia, Planctomycetes, and Bacteroidetes. Soils that have not undergone anthropological actions exhibit an increase ability to degrade carbon sources. The richest soil with the high diversity was found in ecosystems that have suffered anthropogenic action. Our study shows profile of diversity inferring metabolic profile, which may elucidate the mechanisms underlying changes in community structure in situ mining sites in Brazil. Our data comes from contributing to know the bacterial diversity, relationship between these bacteria and can explore strategies for natural bioremediation in mining areas or adjacent areas under regeneration process in iron mining areas.
Brazil is one of the largest producers of iron ore along with China and Australia. Iron ore is a raw material used in the manufacturing of steel. A metal supplies the steel industry and is used in a variety of consumer products. Brazil has extracted 320 million tons of iron ore accounting for almost 10% of world production and feature extraction occurs in rocks and further processing of the ore because this material is mixed with other elements.1 Increasingly, this feature has been exploited in the wild because of global demand, which is, in turn, controlled by population growth and intensification of industrial sectors. Mining activity is causing environmental change in the landscape, particularly by changing the vegetation cover and affecting soil and water from the mine boundary region.2 Furthermore, the mining activities require an additional area to be used for disposal of waste, and years of intensification affect the quality of surrounding soils and aquifers and threaten human health and ecosystems with serious environmental impact.3–5
Many mining companies are aware of the detrimental effects of mining. Companies have invested research and technology into how to practice sustainable exploitation and environmental protection, but these efforts have been associated with an economically less costly production chain. In mining-contaminated regions, soil plays an important role in the transport and storing of contaminants. These characteristics determine the ecological balance and biodiversity of the ecosystem.6 There is a consensus in the literature that metal-contaminated soil exhibits an extremely complex and well-adapted microbial community.7,8 It should be noted that these communities play an important role in biogeochemical cycling and are involved in the transformation of nutrients such as N and C.8
From an environmental standpoint, mining practices are considered unsustainable due to the removal of natural resources until its total depletion (i.e., mineral resources are nonrenewable) and the high negative impact on ecosystems, especially on soil habitats, with the removal of soil layers.9 Procedures to minimize the impacts of mining practices might include the maintenance of vegetation cover as well as preservation of soil macro and microfauna. Therefore, the use of practical tools (i.e., bioindicators) becomes crucial when evaluating biological processes and patterns that may indicate the current environmental condition of soil systems previously subjected to mining activities.10
Considering that microorganisms play an essential role in environmental biogeochemical cycling and may influence the speciation and bioavailability of metals, it is relevant to obtain more comprehensive knowledge of the taxonomic qualities and diversity of the prokaryotic community in metal-contaminated soil.11,12 Although previous studies of the microbial community in metal-contaminated soil have been performed,3,13 none of them have assessed the microbial community of a preserved mine area with five different soils, through taxonomic diversity and functional inference evaluation.
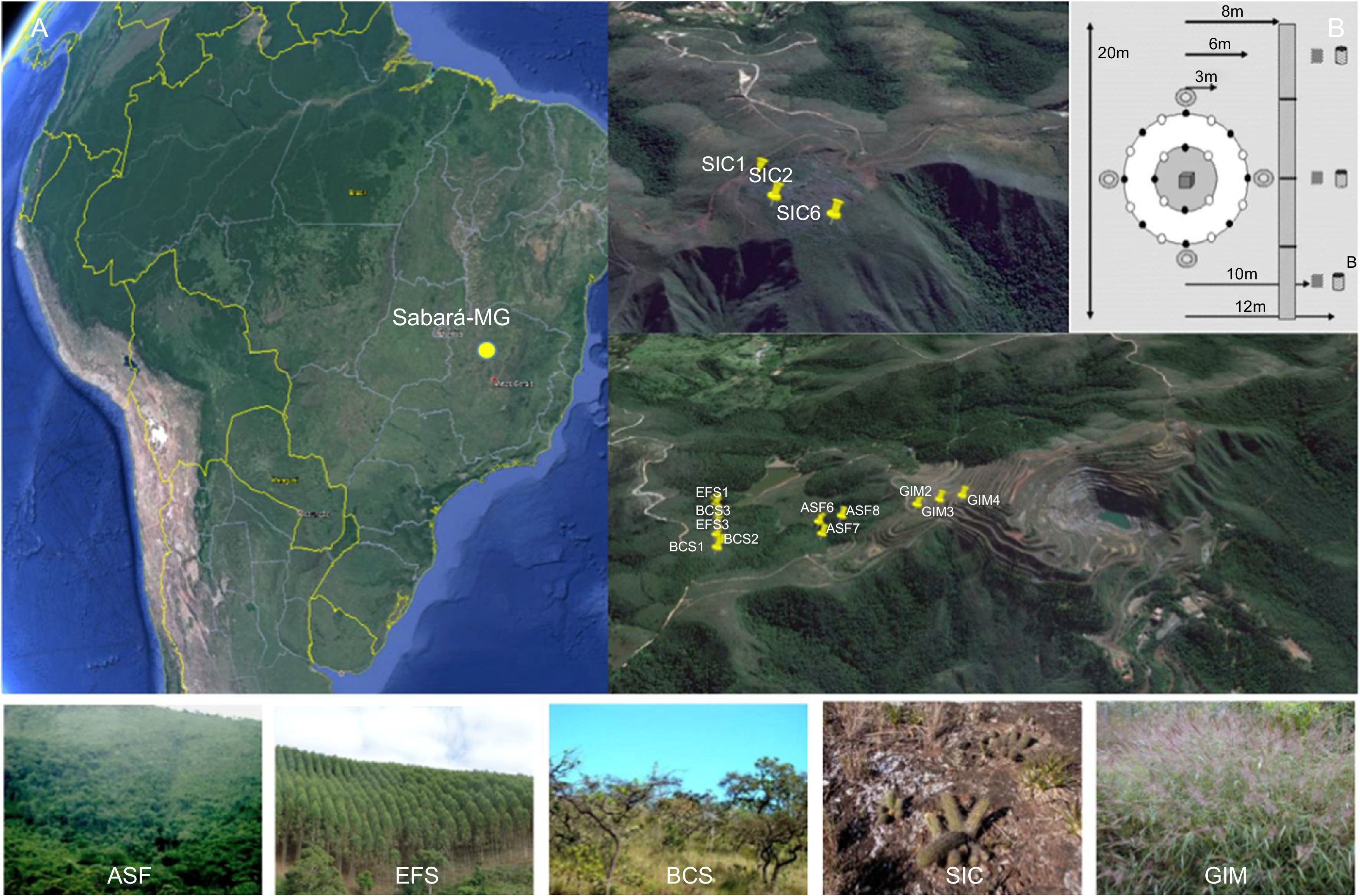
In this study, we assess the current environmental status of an area (Córrego do Meio Mine) subjected to several decades (1946–2005) of mining activities. During this period, deeper layers of soil were removed for the extraction of iron in the area. The removed subterranean soil was accumulated on the surface forming piles of soil next to the open-pit (Fig. 1). In 2006, however, the Center for Research and Biodiversity Conservation of the Iron Quadrangle (CeBio) was established and the area is now under recovery. The CeBio comprises three different types of natural vegetation (i.e., rainforest, savannah and ironstone outcrops). Additionally, two impacted areas are currently under recovery with the introduction of input vegetation.
. Characteristic vegetation of each ecosystem (ASF, EFS, BCS, SIC and GIM) is shown in Table 1. B. Sampling scheme used for collecting soils samples according to Moreira et al. (2010) is shown in the higher right; black point represents the soil samples.")
Location of the Córrego do Meio mine, Sabará, Minas Gerais State, in the southeast of Brazil. A. Sampling site distribution within the different five ecosystems is shown in the enlarged central image (modified from Google Earth). Characteristic vegetation of each ecosystem (ASF, EFS, BCS, SIC and GIM) is shown in Table 1. B. Sampling scheme used for collecting soils samples according to Moreira et al. (2010) is shown in the higher right; black point represents the soil samples.
Monitoring soil quality is key to improving strategies for the recovery of mining areas. In this study, we based on 16S rRNA gene sequences using high-throughput DNA sequencing and a metabolic analysis to examine the taxonomic and infer functional composition of the bacterial community in an off mining area, which currently is in recovery, but during 30 years suffered with mining activities without regeneration process. The issue of this work is showing the bacterial diversity of fundamental importance to deepen our current understanding of microbial interactions in mining ecosystems in regeneration. In addition, the metabolic diversity of bacterial community present in mining sites makes these environments ideal for studies of genomes, ecology, evolution, tolerance mechanisms, and the interactions between bacteria and environmental factors.
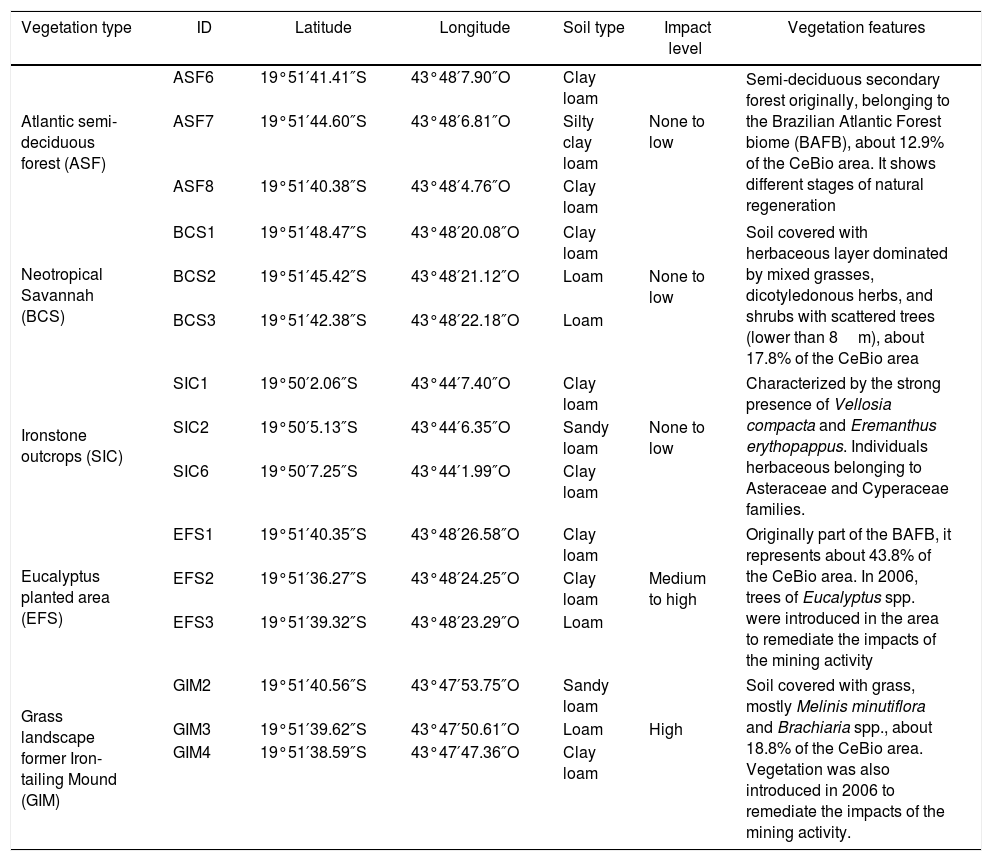
Materials and methodsArea of studyThe study was conducted in the Córrego do Meio Mine area (CeBio), located near the city of Sabará, Minas Gerais State, in the southeast of Brazil (Fig. 1). The mean annual temperature in the region is 20.8°C with a mean rainfall of 1500mm. High yielding iron (Fe) deposits were extracted until mining activities ceased in 2005 due to low production. Based on the vegetation and soil substrate, the mining area (approximately 711ha) is roughly categorized into five ecosystems: Atlantic semi-deciduous forest, neotropical savannah, ironstone outcrops, soil covered with eucalyptus trees and soil covered with grasses.
Soil sampling and identificationSampling was carried out in September of 2013. Three replicate sampling plots were randomly established in the five ecosystems for the collection of soil samples (Fig. 1). These included three natural ecosystems with no level or low level of disturbance: Atlantic Semi-deciduous Forest (ASF), Neotropical Savannah (BCS) and Ironstone outcrops (SIC); and two disturbed areas: Eucalyptus planted area (EFS) and Grass landscape former Iron-tailing Mound (GIM), with medium/high and high impact, respectively (Table 1). The total sampled surface soil (0–20cm in depth) was collected (Fig. 1) and packed in sterile plastic bags. Each bag was sealed and transported on ice to the laboratory for further study. Fractions of the samples were divided in two: one was used to study metabolic diversity and the other was stored at −80°C for molecular analyses.
Ecosystems sampled in this study.
| Vegetation type | ID | Latitude | Longitude | Soil type | Impact level | Vegetation features |
|---|---|---|---|---|---|---|
| Atlantic semi-deciduous forest (ASF) | ASF6 | 19°51′41.41″S | 43°48′7.90″O | Clay loam | Semi-deciduous secondary forest originally, belonging to the Brazilian Atlantic Forest biome (BAFB), about 12.9% of the CeBio area. It shows different stages of natural regeneration | |
| ASF7 | 19°51′44.60″S | 43°48′6.81″O | Silty clay loam | None to low | ||
| ASF8 | 19°51′40.38″S | 43°48′4.76″O | Clay loam | |||
| Neotropical Savannah (BCS) | BCS1 | 19°51′48.47″S | 43°48′20.08″O | Clay loam | Soil covered with herbaceous layer dominated by mixed grasses, dicotyledonous herbs, and shrubs with scattered trees (lower than 8m), about 17.8% of the CeBio area | |
| BCS2 | 19°51′45.42″S | 43°48′21.12″O | Loam | None to low | ||
| BCS3 | 19°51′42.38″S | 43°48′22.18″O | Loam | |||
| Ironstone outcrops (SIC) | SIC1 | 19°50′2.06″S | 43°44′7.40″O | Clay loam | Characterized by the strong presence of Vellosia compacta and Eremanthus erythopappus. Individuals herbaceous belonging to Asteraceae and Cyperaceae families. | |
| SIC2 | 19°50′5.13″S | 43°44′6.35″O | Sandy loam | None to low | ||
| SIC6 | 19°50′7.25″S | 43°44′1.99″O | Clay loam | |||
| Eucalyptus planted area (EFS) | EFS1 | 19°51′40.35″S | 43°48′26.58″O | Clay loam | Originally part of the BAFB, it represents about 43.8% of the CeBio area. In 2006, trees of Eucalyptus spp. were introduced in the area to remediate the impacts of the mining activity | |
| EFS2 | 19°51′36.27″S | 43°48′24.25″O | Clay loam | Medium to high | ||
| EFS3 | 19°51′39.32″S | 43°48′23.29″O | Loam | |||
| Grass landscape former Iron-tailing Mound (GIM) | GIM2 | 19°51′40.56″S | 43°47′53.75″O | Sandy loam | Soil covered with grass, mostly Melinis minutiflora and Brachiaria spp., about 18.8% of the CeBio area. Vegetation was also introduced in 2006 to remediate the impacts of the mining activity. | |
| GIM3 | 19°51′39.62″S | 43°47′50.61″O | Loam | High | ||
| GIM4 | 19°51′38.59″S | 43°47′47.36″O | Clay loam | |||
Soil analyses were conducted in the Laboratory of Soil Analyses at the Universidade Federal de Lavras (UFLA). A standard soil analyses was carried out for each composition sample to determine total concentrations of the following chemicals: K, P, Ca, Mg, Al, Zn, Fe, Mn, Cu, B and S. Additionally, pH, organic matter content and soil texture (clay, silt and sand) were determined. All the soil analyses were performed following the standard protocols of EMBRAPA.14 Cu, Ni and Cr analyses were conducted in the Laboratory of Soil Analyses at the Universidade Estadual Paulista (FCAV/UNESP), concentrations were determined by atomic absorption spectroscopy with air-acetylene flame, according 3050B methodology USEPA,15 determining the concentrations of metals by Melich method 1.16 All statistical analyses were implemented with ANOVA variance and Tukey's test (p<0.05) in software R.
DNA extractionMicrobial DNA was extracted from 0.5-g soil samples using the PowerLyzer® PowerSoil® DNA Isolation Kit (MoBio Laboratories, Solana Beach, CA) according to the manufacturer's protocols.
Bacterial diversity analysesThe V4–V5 regions of the bacterial 16S rRNA were amplified using PCR (95°C for 3min, followed by 35 cycles at 95°C for 30s, 55°C for 90s, 72°C for 45s, and a final extension at 72°C for 5min) using primers17 515F 5′-TGTGNCAGCMGCCGCGGTAA-3′ and 926R 5′-barcode-CCCCGYCAATTYMTT-3′; both primers are degenerate. The barcode is a twelve-base sequence unique to each sample. PCR reactions were performed in triplicate in a 25-μL mixture containing 8μL of KAPA HiFi HotStart ReadyMix (2X) PCR kit (Kapa Biosystems, Boston, USA), 1μL of each primer (10μM), and 50ng of template DNA. Amplicons were extracted from 2% agarose gels and purified using the Zymoclean™ Gel DNA Recovery Kit w/Zymo-Spin™ IC Columns (Zymo Research, Irvine, CA) according to the manufacturer's instructions, and quantified using Qubit Fluorometric Quantitation (Thermo Fisher Scientific, USA). Purified amplicons were pooled in equimolar quantities and single-read sequenced (1x400pb) on an Ion Torrent PGM Sequencer (Thermo Fisher Scientific, USA) on a separate Ion 316 v2 chip, according to the manufacturer's protocols.
Raw data files from sequencing of 16S rRNA gene, region V4-V5, adapter were trimmed using Scythe 0.991 (https://github.com/vsbuffalo/scythe) and Cutadapt 1.7.1.18 The quality of data was filtered by PHRED 20, and duplicate reads were removed using the Prinseq program.19 We used QIIME v. 1.7.020 to filter reads and determine Operational Taxonomic Units (OTUs) at the ≥97% similarity level.21 Briefly, we used the open reference OTU picking workflow, where sequences were first clustered with the Greengenes (GG_13_8_99). Chloroplast-derived contaminant and chimera reads that could not be assembled were removed prior to downstream analyses. For computing alpha diversity metrics among substrates, we used alpha_diversity.py script in QIIME (chao1, phylogenetic diversity and equitability), using the number of the smaller group of samples. We used the beta_diversity_through_plots.py script to compute beta diversity distances between samples (weighted UniFrac) for both the phylogenetic composition and the relative abundance of taxa.
Predicted metabolic profilePredict functional bacterial community profile was performed by PICRUSt.22 OTU table was built with Qiime program (97% similarity) using GreenGenes bank.23 OTUs were normalized in normalize_by_copy_number.py script and metagenomics inference was made by predict_metagenomes.py script.
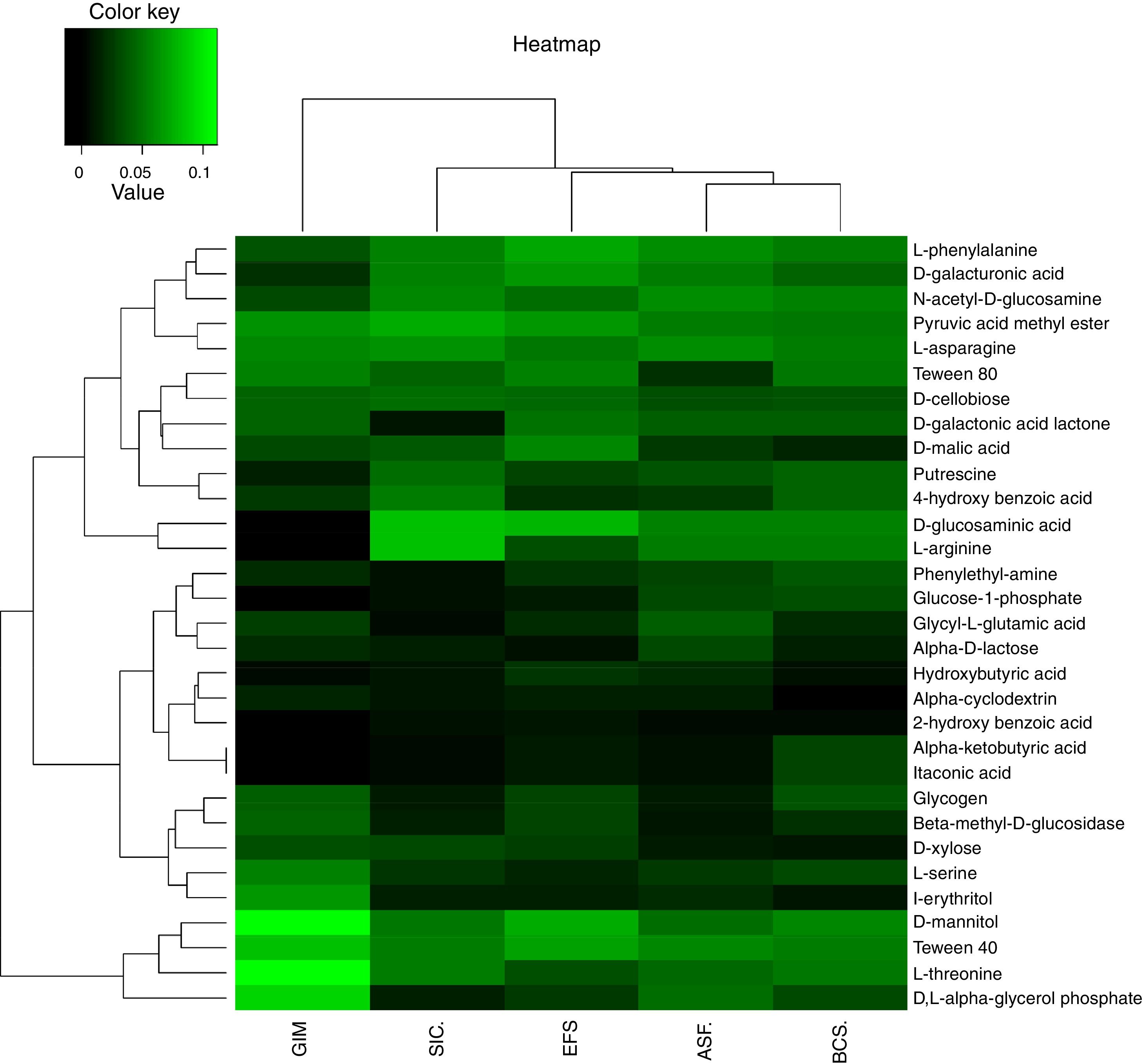
Metabolic diversityBiolog EcoPlates (Biolog, Inc., Hayward, CA, USA) were used to determine the metabolic profile diversity evaluation of microbial populations in the five soils of iron mining area, based on the carbon sources they utilize. The EcoPlate is composed of 31 different carbon compounds divided into six categories along with the control wells in a 96-well microplate (Table S3). Three replicates of each substrate and no-carbon source control were included on a single microplate. Utilization of substrates was spectrophotometrically measured by means of a tetrazolium-formazan reaction, in which colored formazan dye was formed from the tetrazolium salt by the metabolic activity of cells. Ecoplate was prepared in the following way: 2.5g of each sample soil from each region was pooled, and a total of 10g of pooled soil was suspended in 190mL of 0.85% saline solution and shaken for 30min at 30°C. Next, each well of the Biolog EcoPlate was inoculated by 120μL of the prepared suspension and incubated at 28°C.24 Absorbance at 590nm was measured on Bio-Rad Benchmark Microplate Reader after 24, 48, 72, 96, 120, and 144 incubation hours. Optical density (ODi) value from each well was corrected by subtracting the control (blank well) values from each plate well, according to Weber and Legge.25 Optical density values obtained at 144h of incubation represented the optimal range of optical density readings. Thus, we used the incubation results from 144h for the assessment of microbial functional diversity and statistical analyses. Microbial activity in each microplate was expressed as average well-color development, AWCD.26 Hierarchical clustering of each soil was used to assess metabolic diversity.
Cluster analyses were used to evaluate the substrates that were most utilized in each pool of the sample soil. The data were standardized by the average well color development in each microplate to remove inoculum density effects.27 All statistical analyses were performed with software R, version 3.128 and PASTz.29
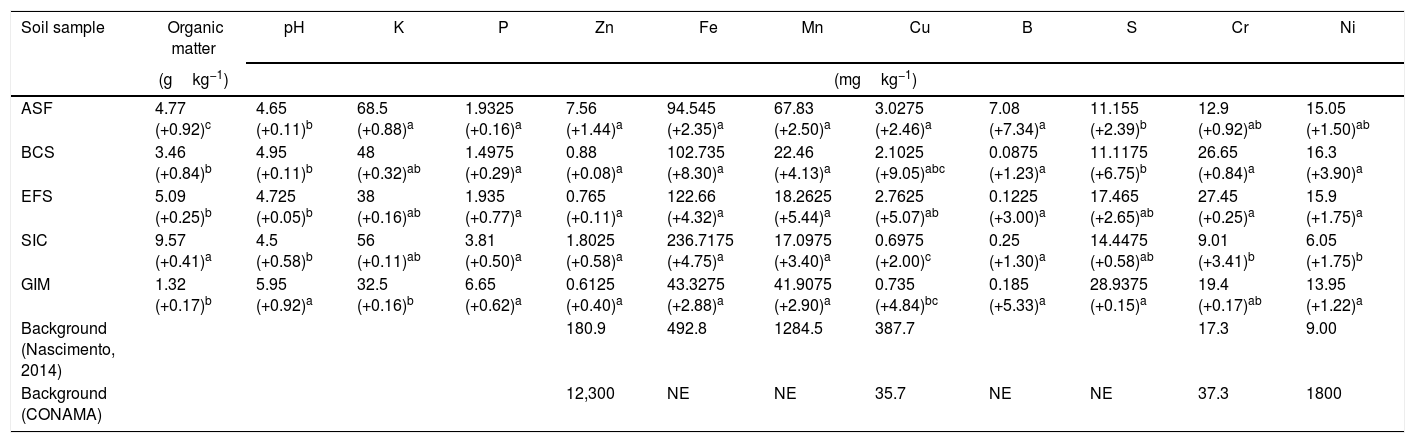
ResultsSoil samples environmental parametersThe chemical characteristics of soil samples from CeBio are presented in Table 2. Data displayed in Table 2 revealed that metal concentration in the CeBio did not exceed the maximum allowable concentrations established by Brazilian environmental regulation30 for soil. Fe, Mn, Cr, and Ni were the metals present in the highest concentrations in the soil samples analyzed.
Chemical parameters and metal concentration from soil samples of CeBio and the limits permitted by law.
| Soil sample | Organic matter | pH | K | P | Zn | Fe | Mn | Cu | B | S | Cr | Ni |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (gkg−1) | (mgkg−1) | |||||||||||
| ASF | 4.77 (+0.92)c | 4.65 (+0.11)b | 68.5 (+0.88)a | 1.9325 (+0.16)a | 7.56 (+1.44)a | 94.545 (+2.35)a | 67.83 (+2.50)a | 3.0275 (+2.46)a | 7.08 (+7.34)a | 11.155 (+2.39)b | 12.9 (+0.92)ab | 15.05 (+1.50)ab |
| BCS | 3.46 (+0.84)b | 4.95 (+0.11)b | 48 (+0.32)ab | 1.4975 (+0.29)a | 0.88 (+0.08)a | 102.735 (+8.30)a | 22.46 (+4.13)a | 2.1025 (+9.05)abc | 0.0875 (+1.23)a | 11.1175 (+6.75)b | 26.65 (+0.84)a | 16.3 (+3.90)a |
| EFS | 5.09 (+0.25)b | 4.725 (+0.05)b | 38 (+0.16)ab | 1.935 (+0.77)a | 0.765 (+0.11)a | 122.66 (+4.32)a | 18.2625 (+5.44)a | 2.7625 (+5.07)ab | 0.1225 (+3.00)a | 17.465 (+2.65)ab | 27.45 (+0.25)a | 15.9 (+1.75)a |
| SIC | 9.57 (+0.41)a | 4.5 (+0.58)b | 56 (+0.11)ab | 3.81 (+0.50)a | 1.8025 (+0.58)a | 236.7175 (+4.75)a | 17.0975 (+3.40)a | 0.6975 (+2.00)c | 0.25 (+1.30)a | 14.4475 (+0.58)ab | 9.01 (+3.41)b | 6.05 (+1.75)b |
| GIM | 1.32 (+0.17)b | 5.95 (+0.92)a | 32.5 (+0.16)b | 6.65 (+0.62)a | 0.6125 (+0.40)a | 43.3275 (+2.88)a | 41.9075 (+2.90)a | 0.735 (+4.84)bc | 0.185 (+5.33)a | 28.9375 (+0.15)a | 19.4 (+0.17)ab | 13.95 (+1.22)a |
| Background (Nascimento, 2014) | 180.9 | 492.8 | 1284.5 | 387.7 | 17.3 | 9.00 | ||||||
| Background (CONAMA) | 12,300 | NE | NE | 35.7 | NE | NE | 37.3 | 1800 | ||||
Tukey analysis – (small letters no difference between ecosystems).
NE – not established.
* CONAMA resolution 420/09.
The physicochemical analyses revealed that except for GIM, pH value from other sites changes slightly from 4.5 to 4.95. Another characteristic from GIM is that it had the highest concentration of phosphorus and sulfate, 6.65 and 28.93mgkg−1, respectively. On the other hand, the organic matter presents the lowest concentration in comparison to the others soils, 1.32gkg−1.
The maximum concentration values of Fe, Mn, Cr, and Ni were observed in SIC, ASF, EFS, and BCS, respectively. Although the Fe concentration was high in all samples, this is not regarded as an indicator of metal pollution in the SIC soil, a native and mined soil from Iron Quadrangle (Table 2).
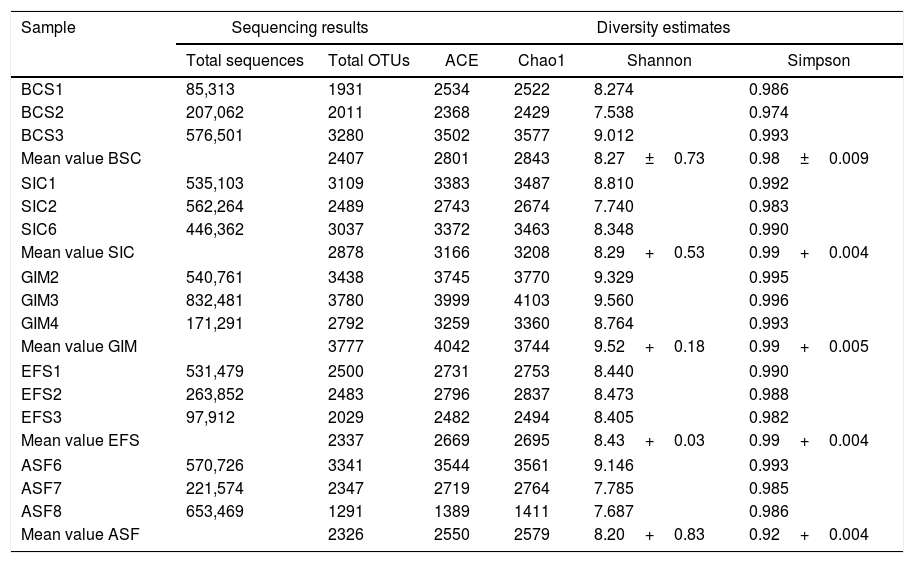
Bacteria diversity analysesThe sequencing of V4 and V5 region of 16S rRNA gene from metagenomic DNA was used to study the bacterial diversity of soils. A total of 8,290,316 reads and 39,858 OTUs of the whole dataset were obtained from fifteen samples through Ion Torrent PGM sequencing analyses. Each library contains 85,313–221,574 total trimmed reads (Table 3). Sequences were trimmed and quality control Phred ≥20 applied. All the results were submitted in Sequence Read Archive (SRA) in NCBI (Table S1). These groups of sequences were aligned in database SILVA.31 We checked the gene region (V4–V5) and used sequences with 0.97 identity cutoff. For statistical analyses, data were normalized using BCS1 (28,750 reads) as a basis, because it was the group that showed less sequence reads (Table 3). Filtering the matrix of distance 0.03 between the sequences (3% different), OTUs for α-diversity were obtained to generate the rarefaction curve, richness (ACE and Chao1), and index diversity (Shannon and Simpson) (Table 3).
Sequencing results and diversity estimates.
| Sample | Sequencing results | Diversity estimates | ||||
|---|---|---|---|---|---|---|
| Total sequences | Total OTUs | ACE | Chao1 | Shannon | Simpson | |
| BCS1 | 85,313 | 1931 | 2534 | 2522 | 8.274 | 0.986 |
| BCS2 | 207,062 | 2011 | 2368 | 2429 | 7.538 | 0.974 |
| BCS3 | 576,501 | 3280 | 3502 | 3577 | 9.012 | 0.993 |
| Mean value BSC | 2407 | 2801 | 2843 | 8.27±0.73 | 0.98±0.009 | |
| SIC1 | 535,103 | 3109 | 3383 | 3487 | 8.810 | 0.992 |
| SIC2 | 562,264 | 2489 | 2743 | 2674 | 7.740 | 0.983 |
| SIC6 | 446,362 | 3037 | 3372 | 3463 | 8.348 | 0.990 |
| Mean value SIC | 2878 | 3166 | 3208 | 8.29+0.53 | 0.99+0.004 | |
| GIM2 | 540,761 | 3438 | 3745 | 3770 | 9.329 | 0.995 |
| GIM3 | 832,481 | 3780 | 3999 | 4103 | 9.560 | 0.996 |
| GIM4 | 171,291 | 2792 | 3259 | 3360 | 8.764 | 0.993 |
| Mean value GIM | 3777 | 4042 | 3744 | 9.52+0.18 | 0.99+0.005 | |
| EFS1 | 531,479 | 2500 | 2731 | 2753 | 8.440 | 0.990 |
| EFS2 | 263,852 | 2483 | 2796 | 2837 | 8.473 | 0.988 |
| EFS3 | 97,912 | 2029 | 2482 | 2494 | 8.405 | 0.982 |
| Mean value EFS | 2337 | 2669 | 2695 | 8.43+0.03 | 0.99+0.004 | |
| ASF6 | 570,726 | 3341 | 3544 | 3561 | 9.146 | 0.993 |
| ASF7 | 221,574 | 2347 | 2719 | 2764 | 7.785 | 0.985 |
| ASF8 | 653,469 | 1291 | 1389 | 1411 | 7.687 | 0.986 |
| Mean value ASF | 2326 | 2550 | 2579 | 8.20+0.83 | 0.92+0.004 | |
All rarefaction curves tended to approach the saturation plateau, indicating the presence of a large variation in the total number of OTUs in different samples (1291–3780 OTUs). Compared soil samples GIM have the highest OTU diversity values; moreover, the calculation of species richness (Chao1), evenness (ACE), and Shannon and Simpson index all confirmed increasing diversity, even in samples in which the total of sequences and OTUs were lower. GIM soil presented a higher value for all indices of diversity: 4042 OTUs for ACE, 9.52 and 0.99 for Shannon and Simpson, respectively. ASF soil had lower values for the same indices, indicating that GIM is an area that is not in balance yet, in recovery, or under construction (Table 3).
Bacterial community composition of CeBioSequences that could not be classified into any known group, Bacteria and Archaea domains, were assigned as unclassified. The bacterial OTUs were assigned into 30 different phyla, 275 families, or 395 genera. Bacteria were by far the most abundant prokaryotic domain. The dominant phyla across all samples were Proteobacteria, Acidobacteria, Verrucomicrobia, and Planctomycetes. However, Bacteroidetes was the most abundant phylum (p<0.001) (Supplement Material S2) in GIM soil compared to the others soils. Although there are differences in the abundance of phylum in all samples, the replicates have the same profile.
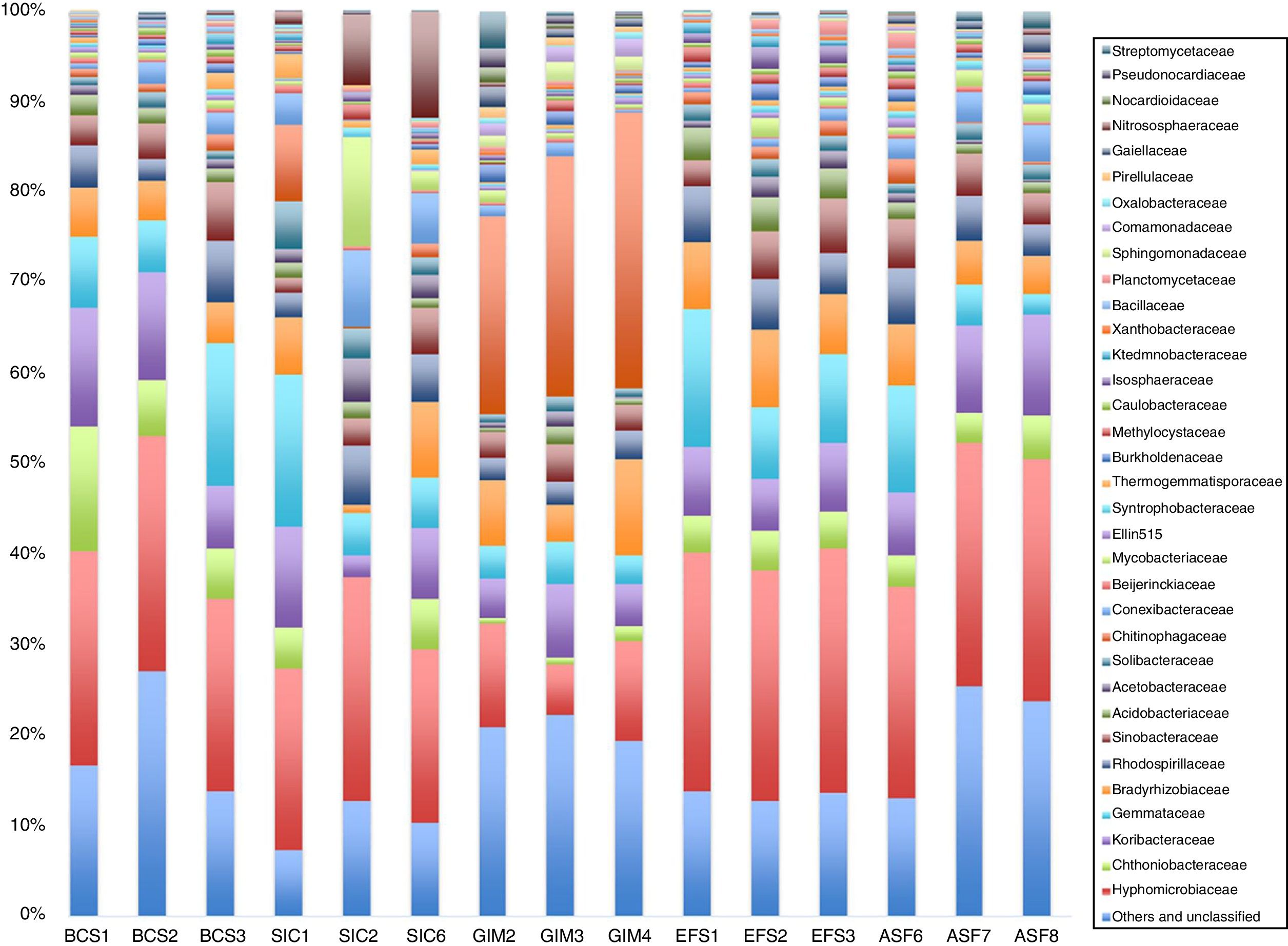
Bacterial family diversity is shown in Fig. 2. Most OTUs were affiliated with Hyphomicrobiaceae, Chthoniobacteraceae, Koribacteraceae, Gemmataceae, and Bradyrhizobiaceae. Similar patterns of bacterial community structure were observed within BCS, SIC, EFS, and ASF soils, while it was different within the GIM soil. The family Chitinophagaceae distribution varied under different soils, it was more abundant in GIM soil when compared to the others. On the other hand, representation of the Chitinoniobacteraceae family was very low in GIM soil when compared to BCS, SIC, EFS, and ASF. The family Chitinophagaceae belongs to the Bacteroidetes phylum, which is consistent with our results for GIM soil samples that had a higher proportion of these bacterium phyla.

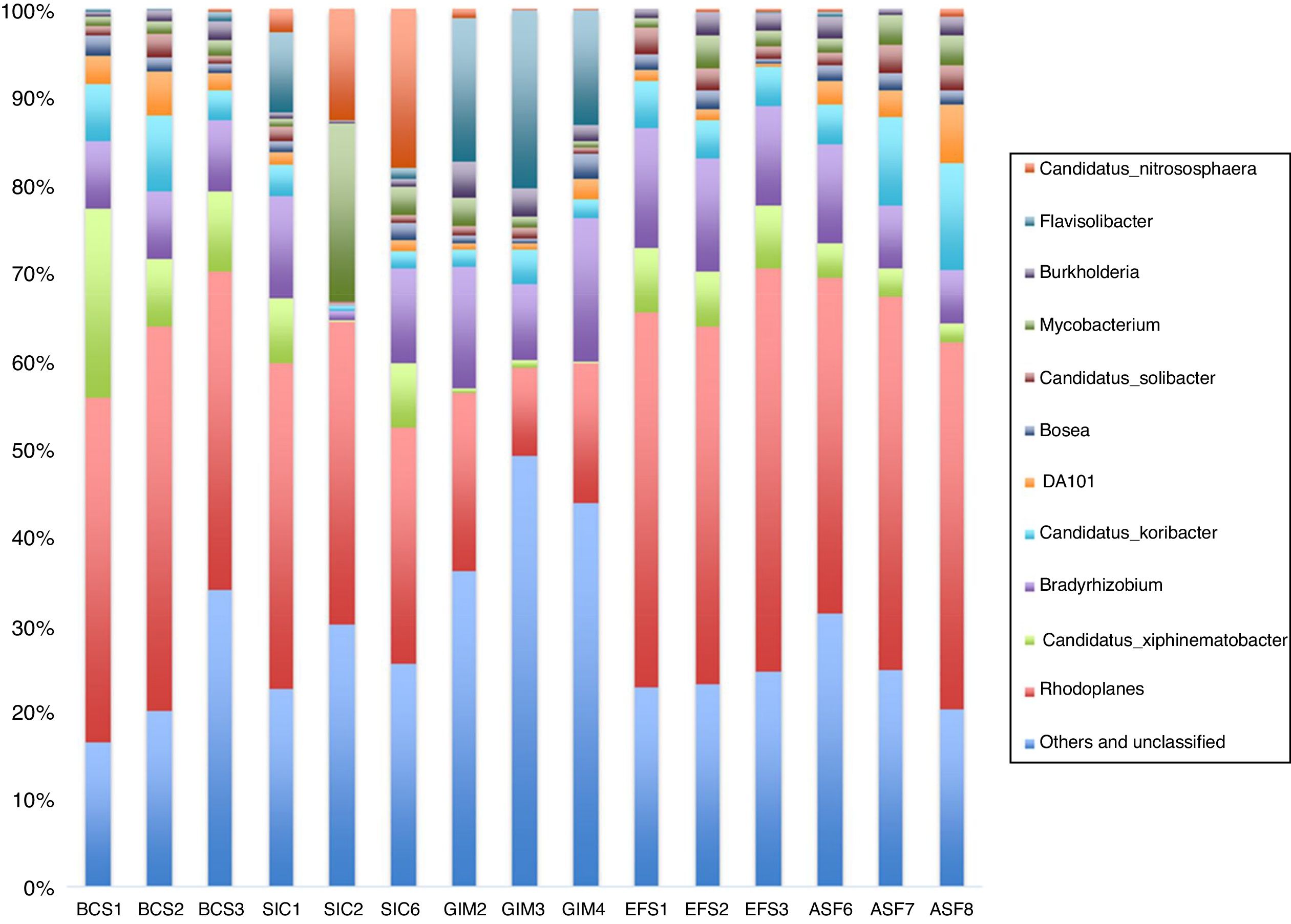
When we analyzed the genera from the bacteria population in each soil (Fig. 3), we can see that the most abundant genera in all samples from CeBio are Rhodoplanes, despite the fact that unclassified and other genera have more than 30% in abundance, mainly in GIM soil. Another finding is that GIM soil had the highest number of representatives from Flavisolibacter genera. The representatives from these genera are Terrimonas, Niastella, and Chitinophaga in the phylum Bacteroidetes.
Predicted metabolic profile
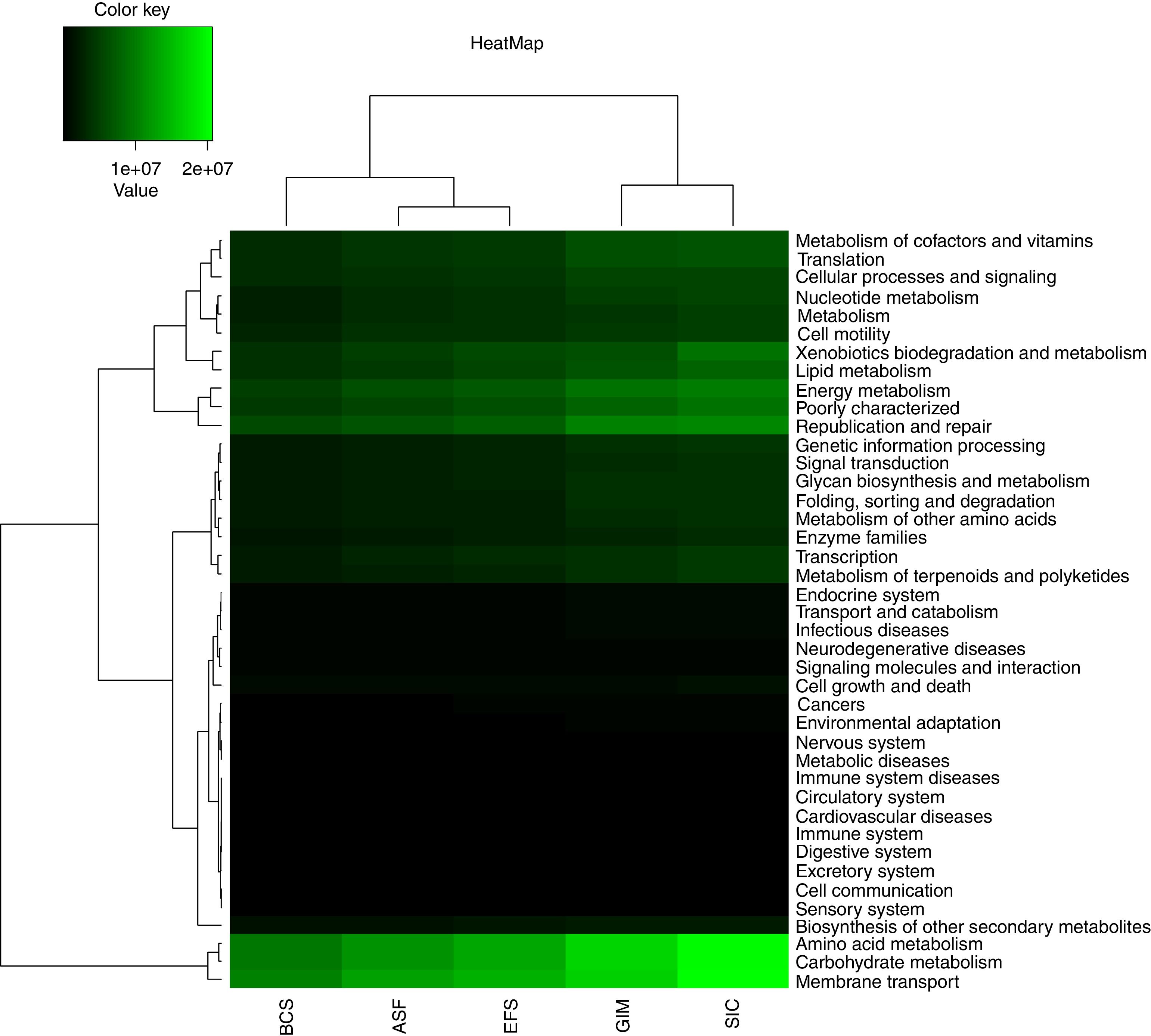
Predict functional diversity from ecosystems was assessed by the abundance of genes identified in PICRUSt in the KEGG database, and we used metabolic via level-2 (Fig. 4). According to this analysis, we can observe a grouping into three distinct groups, one comprising BCS and ASF, non-mined areas from CeBio, the EFS an intermediate group and, the other comprising SIC and GIM native soil and soil mining recovering area from CeBio. There were a few differences in the abundance of level-2 KEGG pathways in ecosystem soils. Only three pathways changed substantially: “amino acid metabolism”, “carbohydrate metabolism”, and “membrane transport”. The most functional diversity for these pathways can be observed in SIC and the lowest in BCS. Others pathways have the same abundance in the different types of soil but in smaller amounts.
Metabolic diversity and community-level physiological profile (CLPP)
The metabolic profile of the bacterial community of CeBio soils was assessed using Biolog Ecoplate (Biolog, Inc.). The list of carbon substrates are shown in Table S3. Carbon substrate utilization by Biolog EcoPlates showed a different metabolic activity in bacterial community from CeBio, the ability of degraded carbon sources in GIM is very different to SIC, ASF, EFS, and BCS (Fig. 5).
 in all time of incubation.")
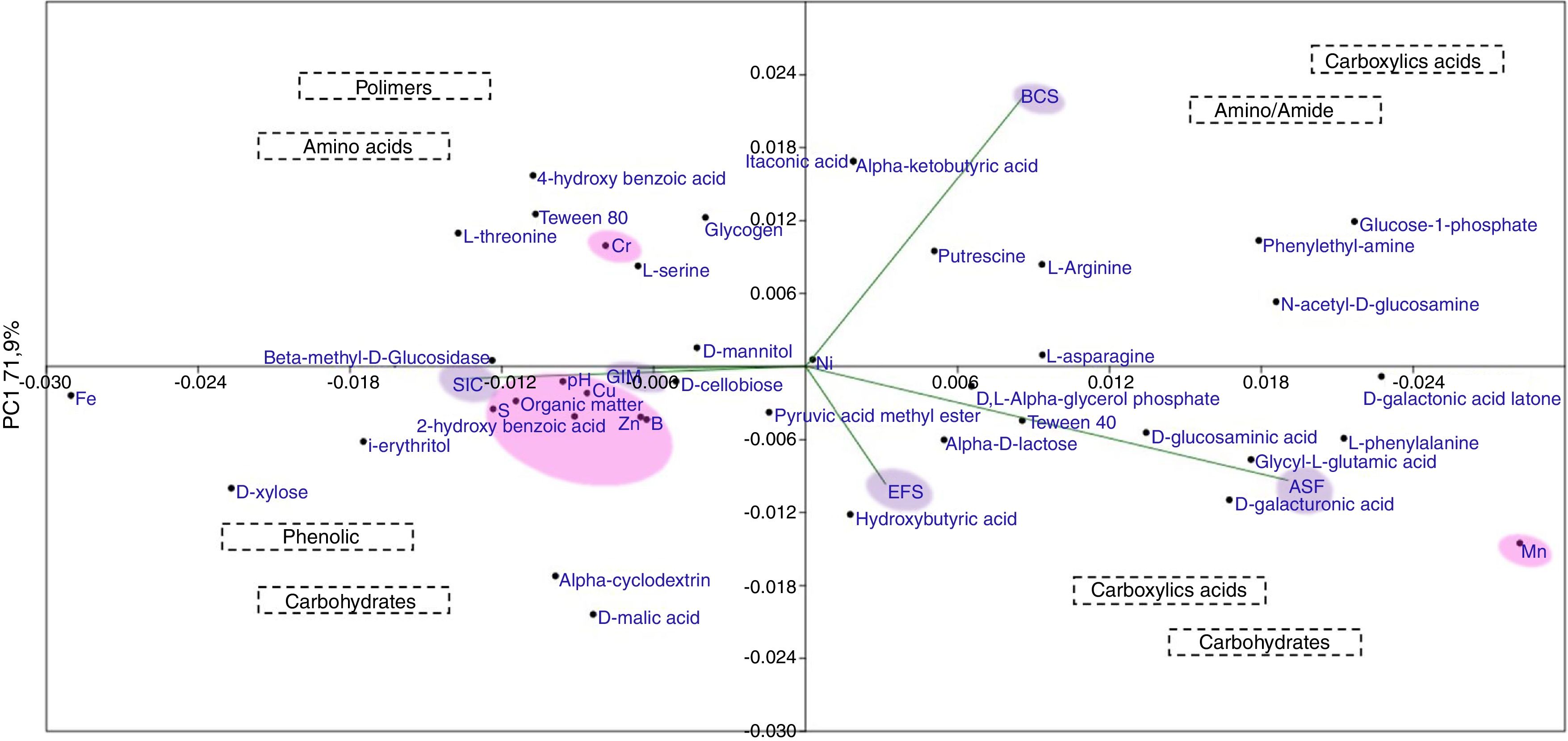
Principal coordinates analyses (PCoA) was used to verify the correlation in the community structure differences under different soil samples. The resolutions of PCoA between physicochemical parameters, metal concentration, carbon source, and ecosystems were different (Fig. 6). Soil samples exhibited different preferences for consumption of carbon sources, highlighting carbohydrates, carboxylic acids, amino/amide, and phenolic compounds. Ecosystems, such as GIM and SIC, showed higher degradation of carbon sources related to carbohydrates and phenolic compounds. Indeed, for ASF, EFS, and BCS, ecosystems displayed carbon source degradation related to carboxylic acids, amino/amide, and carbohydrates. These results demonstrated that bacterial community in GIM and SIC, mining areas, had significant differences in degradation of carbon sources compared to ASF, EFS and BCS, areas that were not mined.
Discussion calculated from the consumption of carbon sources of microbial communities, soil samples, and metals.")
The environmental impact of mining is currently a major concern because its processes release toxic metals, such as Fe, Mn, Cr, and Ni, in soil. One of the most persistent and complex environmental issues is metal pollution, because of threat to ecosystems and human health. In Brazil and in other parts of the world, metals are raw materials used for the manufacturing of steel. Iron ore is the most widely used metal, which supplies the steel industry aimed at a variety of consumer products based on this ore. On the other hand, the companies that explore these areas should practice high control of these mining areas to avoid contamination in the soil, sediment, and water.
Microorganisms constitute the oldest members of living systems and possess higher adaptability to thrive in adverse conditions. The microbial community plays an important role in the soil system, especially in extreme ecosystems where they are responsible for the majority of organic matter decomposition. The dataset presented in this study is a taxonomical and functional characterization of the microbial community in one of the largest mining regions (CeBio), using metagenomics approach.
Taxonomic analyses revealed that a highly complex bacterial community was present in CeBio soils. Taxonomic data indicated that Proteobacteria was the most abundant division, comprising approximately 50% of the bacterial community in all soils. This finding is in accordance with other studies on bacterial community soils,32–34 in which the most dominant was noted to be Proteobacteria. These authors concluded that Proteobacteria could represent 25–40% of total sequences by clone library studies or 42–50% obtained by next generation sequencing. Lieber et al.,35 summarized that, α-, β-, and γ-Proteobacteria, Actinobacteria, Acidobacteria, and to a lesser extent Firmicutes, Bacteroidetes, and Plantcomycetes have been identified as major phyla; however, recognizing that their relative abundances and presence in various types of soils. Studies suggest that the abundance of the Actinobacteria and Firmicutes phyla is significantly correlated with metal-contaminated environments.36–38 However, do not have metal contamination found in CeBio soils, because of it these phyla do not appear in abundance.
In a work with Neutral Mine Drainage Channel, Pereira et al.,39 showed that the phylum Proteobacteria was the most abundant followed by Acidobacteria and Actinobacteria in areas with high amount of copper and that pH influenced Shannon and Simpson indices showing higher values, suggesting less dominance and greater diversity in the alkaline environments, revealing that there was no significant difference between the drainage and soil samples in terms of diversity.
Our results, both soil pH and soil total carbon content dominantly affected the composition and diversity of the soil's bacterial community, and the effect of soil pH was stronger than that of soil carbon content. The pH is an integrating variable indirectly influencing microbial community structure. The reason of the highest pH value, concentration of phosphorus and sulfate and the lowest organic matter in GIM soil, indicating a high environmental heterogeneity. This heterogeneity can be attributed to the differences in time of mining disposal, local availability of air and water, and the levels of nature of microbial metabolic activities of the different sampling sites. Changes in soil management, including land coverage, may lead to changes in energy resources, thus influencing the abundance, diversity and trophic composition of soil microorganisms.40,41
In forest soil, Rodrigues et al.,42 described that the conversion of a forest into arable soils the local taxonomic and phylogenetic diversity of soil bacteria increases after conversion, but communities become more similar in space. This homogenization is driven by the loss of forest soil bacteria with restricted (endemic) bands and thus results in loss of diversity.
The positive relationship between the abundance of Actinobacteria and Bacteroidetes and soil pH have been reported in different soils,43,44 but were not observed in CeBio soils, unless in GIM samples, where Bacteroidetes are present in high abundance. Lauber et al.,43 also found a positive correlation between the pH of the substrate and the relative abundance of Bacteroidetes. In different studies of bacterial communities in soil Bacteroidetes phylum have been found in cultivated fields,38,43 greenhouse soils,44 and unexploited areas.45–47
Environmental Bacteroidetes are specialized in the degradation of complex organic matter,48 they are versatile in a range of biopolymers that they can use as carbon and as an energy source; these biopolymers include plant, algal, or animal compounds. The description of new taxa often results from screening of environmental samples to discover original enzymatic activities with potential biotechnological applications.49–51
Our results indicated that Bradyrhizobiaceae and Rhodospirillaceae are present in high amounts in CeBio soils, mainly in GIM samples, where the soil is predominant covered with grass, mostly Melinis minutiflora and Brachiaria spp, and these families are members of nitrogen-fixing and photosynthetic bacteria. It has been suggested that native nitrogen-fixing plant management combined with nitrogen-fixing bacteria could be an effective measure for the recovery of a degraded ecosystem.52 These ideas can be convenient for re-vegetation in the CeBio mining area; the success of creeper legumes, such as Dolichos lablab, Vicea sativa L, Canavalia ensiformis DC, Desmodium ovalifolium, Arachis pintoi, Lotononis bainesii, Stylosanthes spp., can thus be largely attributed to their ability to form a nitrogen-fixing symbiosis. Because of the bacterial action of absorbing, oxidizing, decomposing, and reducing pollutants, bacteria had a significant contribution on ecological health and soil purification.
The microbial community physiological profile analyses based on the ability to use different carbon sources has been successfully used to characterize microbial diversity in different environments.53–55 Soils under stress and their previously adapted microbiota are an alternative to areas of recovery studies using bioremediation or phytoremediation processes and toxic compounds released to the environment because toxic compounds are harmful to the health of living organisms.56–58 Our data showed a high metabolic diversity in the CeBio soils, the differences in the abundance of microorganisms observed by richness and diversity indices arise with a possible application in bioremediation or phytoremediation. Furthermore, other studies have also reported that in mining areas showed a wide range of prokaryotic.13,59–61
During the Fe extraction period when the mine was active, deeper soil layers were deposited on the surface. Additionally, the natural vegetation (ASF) in this site was entirely removed with mining activities. The introduction of grass vegetation on the almost sterile soil in the GIM may have primarily facilitated the re-colonisation and increase of the abundance of the Bradyrhizobiaceae and Rhodospirillaceae, environmental stressors can also result in a dominance of these bacterial communities when natural vegetation is replaced by agriculture cultivars, and especially when cultivation is in monocrops (e.g., grassland in this study).52
The predictive functional profiling of the bacterial communities of CeBio soils showed that “metabolism” was the most dominant category, of which “amino acid metabolism”, “carbohydrate metabolism” and “membrane transport” were the top three abundant pathways. In the “amino acid metabolism” pathway, which is commonly used by bacteria for osmoregulation, soils have been exposed to frequent moisture stress, mainly GIM area.62,63 “Carbohydrate metabolism” involves a complex series of enzymatic steps to convert external substrate into metabolic precursors, such as acetyl-CoA, pyruvate, and d-fructose-6-phosphate, and it involves a hydrolysis reaction in which polymers are hydrolyzed to monomers.64,65 The “membrane transport” pathway involves an efflux of the irritant metals outside the cell by transporters, transformation of the metals into non-toxic/less toxic forms, and biosorption. Often, biosorption and enzymatic conversion of metal into another form occur at the same time; a metal may be absorbed into the cell, which precipitate the metal as salt.61,64,66
The spatial distribution of soil bacterial communities is strongly linked to physical properties of the soil. We found that the spatial distribution of bacterial communities’ assemblages seems to be determined by a combination of factors such as soil properties, type of subsystems/vegetation as well as impact level, and geographic distribution of samples. For example, within BCS area, samples were closely associated with ASF, as well as SIC and GIM.
In relation to vegetation coverage, noted that almost all high bacteria taxa diversity and intense metabolic profile were more abundant in areas no covered by vegetation when compared with similar areas with vegetation, showing that bacterial community is well adapted to environmental under stress, suggesting a transition from soils with sparse grass vegetation (GIM) to those of dense wooded areas (ASF, BCS and EFS), observed in the PCoA analyzes.
On the other hand, areas re-covered with Eucalyptus spp. have positively affected bacterial composition, resulting in similar characteristics to those found in natural areas. This fact infers that EFS area did not differentiate from natural and none/low impact area, but did differentiate from GIM area (high impact). Altogether, these findings suggest a possible strategy toward recovery EFS to previous rainforest conditions.
ConclusionThe study of microorganisms by metagenomics is a transition from classical microbiology to modern one. This new area is likely to be an important contributor to solve different problems. Metagenomics is a new branches of knowledge and specialization.67,68 In conclusion, our data reveal that the microbial communities from CeBio soils have significantly different features that depend on the particular areas that have undergone anthropological actions, including mining activities (GIM and EFS), from areas that have not experienced anthropological actions (SIC, ASF and BCS). This study provides important insights into the structure of the prokaryotic community of a mining and surrounding area under regeneration process indicating a possible role for this community. The inference of functional diversity analyses unveiled a high degree of diversity, indicating that this microbial community is well adapted to environments under stress, like GIM soil. Finally, the results reported here expand the current knowledge of the microbial taxonomic in mining areas; our data may be a relevant contribution to the exploration of natural bioremediation or phytoremediation strategies in mining and surrounding areas under regeneration process in a former iron mine.
Conflicts of interestThe authors declare no conflicts of interest.
Funding informationThis work was supported by Research Projects for Technologic Innovation – PITE – FAPESP/FAPEMIG/FAPESPA/VALE S.A., n° 01/2010 (grants: 2010/51316-0; 2012/20022-6; 2014/14234-6).
We gratefully acknowledge Universidade Estadual Paulista – UNESP and Universidade Federal de Lavras – UFLA for soil sample analysis, supported by Department of Soil Science. We would like to thank the Programa de Pós Graduação em Microbiologia Agropecuária, UNESP, Jaboticabal, São Paulo State, Brazil. We also thank Universidade Estadual Paulista – UNESP, Departamento de Tecnologia, Laboratório Multiusuário Centralizado para Sequenciamento de DNA em Larga Escala e Análise de Expressão Gênica – LMSeq for sequencing (grants: FAPESP 2009/53984-2).
These authors contributed equally for the paper.
The data sets results of this article are available in the NCBI BioProject PRJNA290860 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA290860).