


We report here the draft genome sequence of Acinetobacter sp. Strain V2 isolated from the oil contaminated soil collected from ENGEN, Amanzimtoti, South Africa. Degradation of phenolic compounds such as phenol, toluene, aniline etc. at 400ppm in 24h and oil degrading capability makes this organism an efficient multifunctional bioremediator. Genome sequencing of Acinetobacter spp. V2 was carried out on Illumina HiSeq 2000 platform (performed by the Beijing Genomics Institute [BGI], Shenzhen, China). The data obtained revealed 643 contigs with genome size of 4.0 Mb and G+C content of 38.59%.
Acinetobacter spp. have been involved in bioremediation of various pollutants such as phenols, benzoate, crude oil, acetonitrile etc.1,2 and biotechnological applications like production of extra-and-intracellular lipases, proteases, bio-emulsifiers and various types of biopolymers.3,4 Physiological and genetic characterisation of large number of phenol-degrading bacteria isolated from various sources have been done,5,6 however, little information on bacteria with a high phenol tolerance and high metabolising activity is available.7 Substrate inhibition and low degradation rate are the key factors which limits their applications.8Acinetobacter sp. V2 strain is able to degrade 400ppm of phenol within 24h.9 Presence of commercially important proteins apart from its ability to degrade diesel and engine oil makes this organism unique and thus genome sequencing.
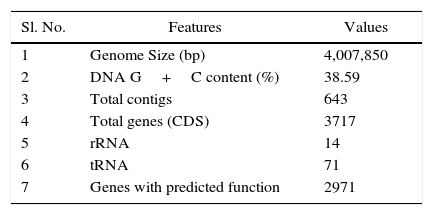
Whole-genome sequencing was carried using Illumina HiSeq 2000 platform (Beijing Genomics Institute [BGI], Shenzhen, China) by generating paired-end libraries with an average insert size of 500bp following the manufacturer's instructions. The reads were then aligned with the reference sequence using SOAPaligner (version 2.21) software to calculate average depth and coverage ratio (http://soap.genomics.org.cn/soapaligner.html, version 2.21).10 The filtered short reads were first de novo assembled using SOAPdenovo v 2.04 according to the method described previously (http://soap.genomics.org.cn/soapdenovo.html),11 and then contigs were manually connected according to their 500bp paired-end relationships. The draft genome sequence of Acinetobacter sp. V2 strain comprised of 643 contigs and 16 scaffolds with the maximum contig size of 910,143bp and scaffold size of 1,913,879bp (Table 1). The genome size was 4,007,850bp at 109.2×coverage, with N50 of 466,223bp and N90 of 97,271bp and G+C content was 38.59%. A total of 3717 coding sequences (CDSs) or ORFs were predicted using Glimmer v3.02,12 and homologous comparison to a non-redundant public database was performed by BLAST for function annotation. The genome annotation was performed using the (BASys) server (https://www.basys.ca/) and the output was downloaded in GenBank format resulting in 3742 (CDSs).13 The genome was further annotated with Rapid Annotation using Subsystems Technology (RAST) server (http://rast.nmpdr.org/).14 Among the predicted 3742 protein-coding genes by BASys, 79.4% (2971genes) have been assigned putative functions according to the subsystem categorization. A total of 71 tRNA genes encompassing all 20 amino acids were identified using the tRNAscan-SE program,15 14 rRNA genes were identified using RNAmmer16 and the insertion sequence (IS) elements were annotated by ISsaga.17 All the contigs were submitted to the Gene bank and NCBI has published sequence data in April 2015. The further analysis is going on.
Nucleotide sequence accession numbers: This WGS project has been deposited at DDBJ/EMBL/GenBank under the accession JZFB00000000. The version described in this paper is version JZFB01000000. Bioproject registered under accession: PRJNA275383 ID: 275383. The Acinetobacter sp. V2 isolate was deposited at the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures and is available under the Accession No. DSM 101893.