





In this study, the development and assessment of a modified, efficient, and cost-efficient protocol for mDNA (metagenomic DNA) extraction from contaminated water samples was attempted. The efficiency of the developed protocol was investigated in comparison to a well-established commercial kit (Epicentre, Metagenomic DNA Isolation Kit for Water). The comparison was in terms of degree of shearing, yield, purity, duration, suitability for polymerase chain reaction and next-generation sequencing in addition to the quality of next-generation sequencing data. The DNA yield obtained from the developed protocol was 2.6 folds higher than that of the commercial kit. No significant difference in the alpha (Observed species, Chao1, Simpson and PD whole tree) and beta diversity was found between the DNA samples extracted by the commercial kit and the developed protocol. The number of high-quality sequences of the samples extracted by the developed method was 20% higher than those obtained by the samples processed by the kit. The developed economic protocol successfully yielded high-quality pure mDNA compatible with complex molecular applications. Thus we propose the developed protocol as a gold standard for future metagenomic studies investigating a large number of samples.
Recently, the evolution of the discipline of “Metagenomics” has grasped considerable attention worldwide with tremendous expectations.1 Several ecosystems are still untapped and will be studied and explored through metagenomic studies. This has been facilitated by the vast advances in molecular tools such as the next-generation sequencing (NGS).2,3
However, an important prerequisite for a successful metagenomic analysis is the use of an efficient protocol for extracting whole community DNA from environmental samples.4 Therefore, a mDNA (metagenomic DNA) extraction protocol has been considered to be the milestone and the main gate to accurate, reliable, and successful metagenomic analyses.5 Accordingly, the isolation of high-quality DNA that covers the whole microbial diversity in the original sample was phrased as a limiting step for the construction of metagenomic libraries and other DNA based metagenomic projects.6
In these regards, many studies have reported the development of methods and even commercial kits for mDNA extraction.5,6 However, still, there is a surge in developing, improving, and optimizing new or current protocols for mDNA extraction from different types of samples. A metagenomic DNA extraction must be followed by a quality control of the extracted DNA. Despite the recent advances in the molecular tools, many researchers still use relatively conventional molecular techniques such as PCR, restriction digestion, and cloning for testing the quality of the extracted mDNA.7–9 In this perspective, the current study developed a modified protocol for the extraction of mDNA from contaminated water samples with the introduction of NGS as a quality control tool to measure the efficiency of the extraction protocol and the quality of the extracted DNA. The quality of the extracted DNA was compared to that of a well established commercial kit in terms of degree of shearing, yield, purity, time requirement, suitability for PCR and NGS, and the quality of NGS data.
Materials and methodsSamples collections and preparation for DNA extractionIndustrial wastewater from a coal coking factory in Egypt (sample A) was used in this study. The samples were collected from the biological wastewater treatment plant at the factory. Other water samples (13 samples, named as B–N) were also collected from a photo-bioreactor using an algal-bacterial system to treat coking wastewater.
The sufficient volume of the samples was calculated according to a predetermined relationship between the dry weight of biomass and the sample volume to reach a final biomass dry weight of 1.7mg for the DNA extraction. These volumes were immediately filtered using a sterile filtration unit (Glassco®, India) and vacuum pump (Pro-set, CPS®, Germany). The samples were filtered using sterile 0.2μm pore size cellulose nitrate membrane filters (47mm diameter, Sartorius®, Germany). The membrane filters were then stored frozen at –20°C till DNA extraction.
DNA extraction using commercial kitMetagenomic DNA isolation kit for water (Epicentre®, USA) was used according to the manufacturer's instructions. The extracted DNA pellets were re-suspended in molecular grade water (DNase and RNase free water) and stored at −20°C till further use.
DNA extraction by the developed approach (chemical protocol)Several trials were attempted to optimize a protocol for the extraction of environmental mDNA. The optimized protocol was achieved through the following steps: a 0.2-μm membrane filter carrying the mDNA was aseptically placed at the center of a sterile 15-mL falcon tube. Then 5mL of extraction buffer (1%, w/v cetyltrimethylammonium bromide (CTAB), 3%, w/v sodium dodecyl sulfate (SDS), 100mM Tris–HCl, 100mM NaEDTA, 1.5M NaCl, pH 8.0) was added to the falcon tube. The resulting solution was incubated in a water bath (65–70°C) for 60min with intermittent vortexing. The content was centrifuged for 15min at 4500×g and the supernatant was transferred into a new sterile falcon tube. Isopropanol (4mL) was liquated to the supernatant and incubated on ice for 20min. The content was centrifuged at −4°C and 4500×g for 15min and the supernatant was carefully aspirated and discarded. The DNA pellets were washed with 200μL of 70% ethanol and centrifuged at −4°C and 4500×g for 10min. Again the supernatant was discarded by careful aspiration and the DNA pellets were fully dried in a laminar flow cabinet. The dried pellets were re-suspended in 100μL molecular grade water (DNase and RNase free water) and stored at −20°C till further use.
Further purification of the extracted mDNAThe previously mentioned protocol was used for further purification of the extracted mDNA using gel purification technique. Where 25μL of the extracted mDNA was resolved on an agarose gel in 0.8% TAE buffered agarose gel and visualized using ethidium bromide. DNA band was then sliced (Supplementary Fig. S1-A) and purified using UltraClean® 15 DNA Purification Kit (Mo Bio®, USA) according to the manufacturer's instructions.
DNA detection by gel electrophoresis and nanophotometerThe size and degree of shearing of the extracted DNA were validated by comparison to a fosmid control DNA with a size of 40kb and concentration 100ng/μL (provided in the Metagenomic DNA Isolation Kit for Water, Epicentre). The fosmid control DNA and the extracted mDNA were electrophoresed in 0.8% TAE buffered agarose gel and were visualized using ethidium bromide.
The quantification of the extracted mDNA was done by a spectrophotometric method using nanophotometer (Implen, NanoPhotometer® P-330, Germany).10 The purity of the extracted DNA was also checked by the absorbance ratios at 260/230nm and 260/280nm calculated using the nanophotometer.10–12
PCR amplificationThe quality of the extracted mDNA was also assessed by its liability to enzymatic reactions using PCR. This also tested the presence of any impurities that might inhibit the enzymatic reactions. Two universal 16S ribosomal DNA primers 28F 5′AGAGTTTGATCCTGGCTCAG-3′ (positions 8–28 in Escherichia coli numbering) and 1512R 5′ACGGCTACCTTGTTACGACT-3′ (positions 1512–1493 in E. coli numbering) were used.13 The positive control employed the genomic DNA extracted from Pseudomonas aeruginosa (ATCC 9027), while the negative control was devoid of any DNA template.
DNA sequencingIllumina-MiSeq™ sequencing (2×300bp paired-end protocol) was performed using the universal primers 341F 5′-CCTACGGGNGGCWGCAG-3′ and 805R 5′-GACTACHVGGGTATCTAATCC-3′ to target the V3 and V4 regions of the 16S rRNA gene.14 The used sequencing kit was MiSeq Reagent Kit V3, 600 cycle (Illumina, USA).
Availability of sequencing dataThe 16S sequence data were submitted to the Sequence Read Archive (SRA) under Bioproject (PRJNA353621) and SRA study (SRP093422). The data have been released by NCBI (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP093422) and assigned the accession numbers: SRR5028842, SRR5028843, SRR5029151, SRR5029152, SRR5029153, SRR5029154, SRR5029155, and SRR5029156.
NGS data analysisIllumina sequencing reads were processed using the software QIIME “Quantitative Insights Into Microbial Ecology”, version 1.9.1 as described in detail in the supplementary data.15 All statistical analyses, unless specified otherwise, were performed using python scripts implemented in QIIME version 1.9.1 (as described in detail in the supplementary data).
ResultsDetection of extracted metagenomic DNA and quality assuranceIn this study, three sets of metagenomic DNA samples, referred to as the kit extracted samples, chemical protocol extracted samples, and gel-extracted samples were considered. The three investigated methods yielded metagenomic DNA with a size in the range of 40kb, compared to the fosmid control (Supplementary Fig. S1-B). The extracted metagenomic DNA showed a single band when resolved on an agarose gel (Supplementary Fig. S1) with no other sheared bands. The humic contaminants were seen in the agarose gel of the metagenomic DNA extracted by both the Epicentre kit and the chemical protocols, yet this humic substance was completely absent in the gel extracted DNA samples (Supplementary Fig. S1-B).
The developed chemical protocol recorded the highest DNA yield of 21±5μg/mg biomass, while the Epicentre kit recorded significantly lower DNA yield of 8±2μg/mg biomass (one way ANOVA, p-value=0.0026) (Fig. 1-A). On the other hand, the metagenomic DNA extraction by gel purification method showed a significantly low yield of 4±1μg/mg of biomass, when compared to the yield of the chemical protocol (one way ANOVA, p-value=0.0026). Yet, there was no significant difference between the DNA yielded by both the Epicentre kit and gel purification method (t-test, p-value=0.1032). The same pattern was observed with the DNA concentrations, where the chemical protocol significantly recorded the highest DNA concentration of 690±180ng/μL (one way ANOVA, p-value=0.0007) (Fig. 1-B). While the lowest DNA concentration (33±9.6ng/μL) was recorded by the gel purification method (Fig. 1-B).
. (1-A) DNA yield expressed as micrograms of DNA per milligram of biomass. (1-B) DNA concentration expressed as ng/μL. (1-C) Absorbance ratios at 260/280nm. (1-D) absorbance ratios at 260/230nm. (*) means that there is a significant difference at p-value<0.05 (One-way ANOVA, Tukey's multiple comparison test).")
Scatter dot plots of comparative analysis of the metagenomic DNA extracted using the three studied methods (kit, chemical protocol, and gel extraction method). (1-A) DNA yield expressed as micrograms of DNA per milligram of biomass. (1-B) DNA concentration expressed as ng/μL. (1-C) Absorbance ratios at 260/280nm. (1-D) absorbance ratios at 260/230nm. (*) means that there is a significant difference at p-value<0.05 (One-way ANOVA, Tukey's multiple comparison test).
The efficiency of removing protein contamination from the metagenomic DNA was validated by the absorbance ratios at 260/280nm and was greater than 1.7 for all three used extraction methods (Fig. 1-C). The recorded absorbance ratios at 260/230nm for the Epicentre kit protocol and the developed chemical protocol were 1.5±0.1 and 1±0.1, respectively (Fig. 1-D). However, the gel purification method showed significantly low absorbance ratios at 260/230nm of 0.1±0.2 (one way ANOVA, p value<0.0001) (Fig. 1-D).
The quality of the extracted metagenomic DNA and its suitability for downstream processing were investigated by using PCR. The metagenomic DNA extracted by the three evaluated methods (kit, chemical and gel purification) showed successful PCR amplification of the targeted sequences of 16S ribosomal DNA (Supplementary Fig. S2).
Metagenomic DNA sequencingIn order to test the suitability of the extracted metagenomic DNA to more challenging and complex downstream processing and further validate the purity and quality of the extracted DNA, the samples were tested for their fitness to Illumina MiSeq™ next-generation sequencing (NGS). Since the DNA extracted by gel purification method resulted in the lowest concentration (Fig. 1-B), this method was excluded from the NGS analysis. Four metagenomic DNA samples (A, B, C, and D) processed by each Epicentre kit and the developed chemical protocol were considered in this investigation (a total of 8 samples).
The number of high-quality sequences observed with the metagenomic DNA samples extracted by the chemical protocol (101,109±3563) was higher than that observed by the Epicentre kit samples (85,026±12,320). However, this difference was not significant (t-test, p-value=0.0711). Again, there was no significant difference between the number of observed OTUs in the metagenomic DNA samples extracted by the developed chemical protocol (1718±365.1) and that extracted by the Epicentre kit (1796±373.2) (t-test, p-value=0.886).
A total of 44 phyla were identified in both the groups of samples (the developed protocol and kit samples), with no significant difference in the relative abundance of each phylum between the two groups of the samples (Kruskal–Wallis test with group significance.py, p-value>0.05) (Fig. 2). The four major bacterial phyla identified in the samples were Proteobacteria (Gram-negative), Firmicutes (Gram-positive), Bacteroidetes (Gram-negative), and Deferribacteres (Gram-negative) (Fig. 2).
 and those extracted by the commercial Epicentre kit (Kit). In the figure each phylum is represented by a circularly arranged ribbon, whose width is proportional to its relative abundance.")
The relative abundance of the identified phyla in the samples extracted by the developed chemical protocol (Chemical) and those extracted by the commercial Epicentre kit (Kit). In the figure each phylum is represented by a circularly arranged ribbon, whose width is proportional to its relative abundance.
The alpha diversity (within community diversity) in the samples extracted by both protocols (kit and chemical protocol) was determined and compared. There was no significant difference (non-parametric Kruskal–Wallis test with compared alpha diversity.py, p-value=0.682) in the observed species richness (number of unique OTUs per number of sequences) between the samples extracted by Epicentre kit and the chemical protocol (Fig. 3-A). The rarefaction curves of the true richness (Fig. 3-A) were compared to the curves of the estimated species richness (indicated by chao1 richness estimator) (Fig. 3-B). The rarefaction curves showing the estimated richness (Fig. 3-B) of both protocols (kit and chemical protocol) were superimposed and no significant difference was recorded between the tested protocols (non-parametric Kruskal–Wallis test with compare alpha diversity.py, p-value=0.962).
 and the commercial Epicentre kit (Ep). (3-A) The observed species indicating the true species richness, (3-B) Chao1 richness estimator indicating estimated species’ richness, (3-C) Simpson's index indicating species’ evenness and (3-D) PD whole tree indicating the phylogenetic diversity.")
Rarefaction curves of alpha diversity measured for the metagenomic DNA samples extracted by the developed chemical protocol (Ch) and the commercial Epicentre kit (Ep). (3-A) The observed species indicating the true species richness, (3-B) Chao1 richness estimator indicating estimated species’ richness, (3-C) Simpson's index indicating species’ evenness and (3-D) PD whole tree indicating the phylogenetic diversity.
Simpson's index rarefaction curves for both methods (kit and chemical protocol) were completely superimposed (Fig. 3-C), with no significant difference (non-parametric Kruskal–Wallis test with compare alpha diversity.py, p-value=0.952). The samples extracted by both methods (kit and chemical protocol) reached a plateau after >16,000 sequences (Fig. 3-C). This indicated that the sequencing depth was successful in capturing the existing microbial diversity in all the investigated samples.
The PD whole tree (alpha diversity measure) was used to represent the phylogenetic diversity between samples extracted by Epicentre kit and the developed chemical protocol (Fig. 3-D). There was no significant difference between the two investigated protocols in terms of PD whole tree (non-parametric Kruskal–Wallis test with compared alpha diversity.py, p-value=0.984).
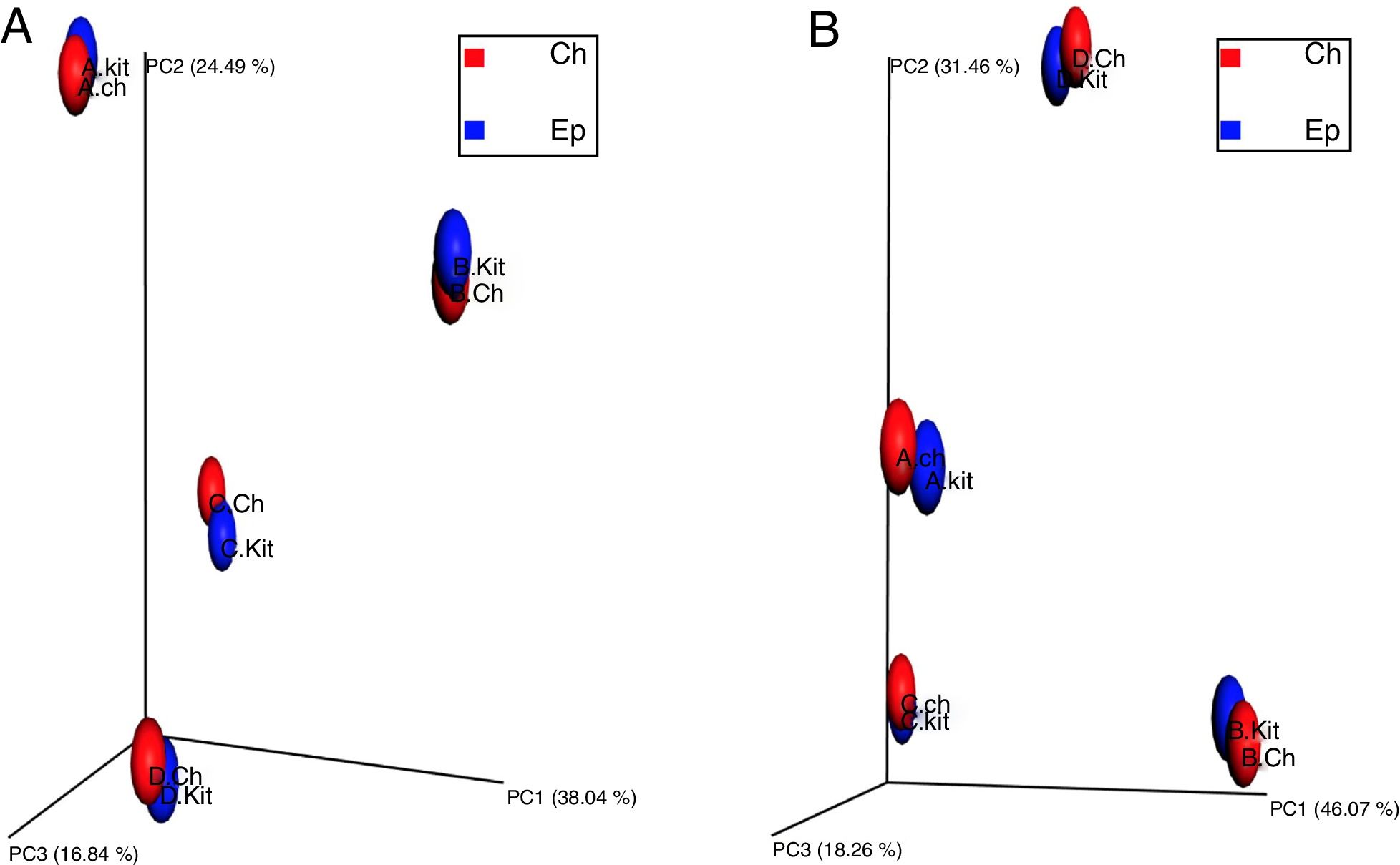
To investigate the beta diversity (between community diversity) between the metagenomic DNA samples extracted by the Epicentre kit and developed chemical protocol, the UniFrac distance metrics were used, as UniFrac takes phylogenetic relatedness into account while computing beta diversity. To explore the differences in overall microbial community composition across the metagenomic DNA samples extracted by the two investigated protocols, both the phylogenetic unweighted UniFrac distances (Fig. 4-A) (consider only the presence or absence of taxa) and weighted Unifrac (Fig. 4-B) (consider taxon relative abundance) were computed. The principal coordinates analysis plot (PCoA), showed that the metagenomic DNA from the same sample source clustered together regardless of the used method of extraction (Fig. 4). Similarly, the beta diversity analysis of the samples showed that each sample source represented a distinct microbiome irrespective of the used DNA extraction method.
 representing the beta diversity estimated by the unweighted UNIFRAC (4-A) and weighted UNIFRAC (4-B) method. Individual datasets are represented as spheres, which are colored according to their sample source as follows; Blue: metagenomic DNA samples extracted by the commercial Epicentre kit, Red: metagenomic DNA samples extracted by the developed chemical protocol. (4-A) The three principal coordinates (PC1–PC3) explain 38.03, 24.49 and 16.84%, respectively. (4-B) The three principal coordinates (PC1–PC3) explain 46.07, 31.46 and 18.26%, respectively.")
Principal coordinates analysis (PCoA) representing the beta diversity estimated by the unweighted UNIFRAC (4-A) and weighted UNIFRAC (4-B) method. Individual datasets are represented as spheres, which are colored according to their sample source as follows; Blue: metagenomic DNA samples extracted by the commercial Epicentre kit, Red: metagenomic DNA samples extracted by the developed chemical protocol. (4-A) The three principal coordinates (PC1–PC3) explain 38.03, 24.49 and 16.84%, respectively. (4-B) The three principal coordinates (PC1–PC3) explain 46.07, 31.46 and 18.26%, respectively.
The significance of the used DNA extraction method on the overall microbial community composition was tested using permutational multivariate ANOVA. There was no significant effect of the metagenomic DNA extraction methods used in the overall community composition within the individual samples (PERMANOVA, p value=0.816 and ANOSIM, p-value=0.813).
DiscussionCoking wastewater is a complex highly contaminated industrial effluent, which has been studied in a great detail.16 Hence, coking wastewater samples from both coal coking factory and photosynthetically aerated bioreactor were selected for this study. The developed chemical protocol for the isolation of metagenomic DNA from the contaminated water samples was compared to a commercial kit (Metagenomic DNA isolation kit for water, Epicentre, USA), as it is a well-established kit that has been used through recent metagenomic studies.17
In the developed protocol, CTAB and SDS were used as purifying agents for the removal of phenolic contaminants and humic substances as described earlier in many DNA extraction protocols.8 The inclusion of purifying agents in the DNA extraction buffer is sufficient to yield DNA with an adequate purity that complies with the different molecular applications. This eliminates the need for further steps with a separate purifying buffer or any additional purification steps as reported by other previously studied DNA extraction protocols.5,9,11 Therefore, this adds extra benefits of the developed protocol in terms of time and money saving.
Although the use of organic solvents (phenol/chloroform) is common in the previously developed metagenomic DNA extraction protocols,18 yet the use of such hazardous solvents was avoided in the protocol developed in this study. Several studies have attempted to enhance the quality and yield of the metagenomic DNA extracted from environmental samples by using physical treatments (bead beating and sonication) in the extraction protocols. However, these harsh treatments usually cause shearing of DNA, which makes it unsuitable for further downstream processing.8 Accordingly, these severe treatments were not included in the developed chemical protocol, yielding un-sheared metagenomic DNA with a size of ≈40kb.
The yield of DNA in the developed chemical protocol was significantly higher (2.6 folds) than that of the Epicentre kit (one-way ANOVA, p-value=0.0026). Similar observations have previously been made in other studies where the adopted chemical protocols yielded higher DNA than the used commercial kits.8 The three protocols used in this study (kit, chemical, and gel purification) achieved sufficient removal of proteins from the extracted metagenomic DNA with the absorbance ratios at 260/280nm>1.8.10,11 However, the highest absorbance ratios (260/280nm) were recorded by the gel purification method, indicating that the metagenomic DNA extracted by this method had the lowest protein concentrations. The metagenomic DNA extracted by the kit and the developed chemical protocol recorded the absorbance ratios at 260/230nm<2, this indicated the presence of humic substances.9,11
Relatively high metagenomic DNA concentrations and reasonable absorbance ratios do not always indicate the suitability of extracted DNA for enzymatic reactions. For instance, a previous study8 investigated five different metagenomic DNA extraction methods but none of the extracted DNA samples was suitable for PCR and/or restriction digestion and the samples required further purification steps. Therefore, many studies have used PCR in the quality assessment of the metagenomic DNA, as positive PCR results indicate the absence of any impurities that may inhibit enzymatic reactions.8,11 In this regard, the metagenomic DNA extracted by the chemical protocol developed in this study showed positive PCR results emphasizing the efficiency of the developed protocol.
The development of a sensitive, reproducible, and precise metagenomic DNA extraction protocol is essential for a successful metagenomic analysis. Many earlier studies have used different molecular techniques as PCR, restriction digestion, and cloning for testing the quality of the extracted metagenomic DNA,8,9,11 but none reached the extent of testing the quality of the extracted DNA by applying the downstream analysis of the NGS output data. Besides, metagenomic DNA extraction protocols intended for the application in NGS require high-quality standards. Specifically, NGS requires reproducible precise protocols that provide adequate read length, number of sequences per sample, and consequently the high quality of NGS data. In this perspective, the current study adopted an initiative approach to applying NGS as a quality control tool for developing and optimizing the metagenomic DNA extraction protocol.
Interestingly, the number of high-quality sequences obtained from the metagenomic DNA samples extracted by the chemical protocol was 20% higher than that of the samples extracted by the Epicentre kit. This occurred as the concentration of DNA extracted by the chemical protocol was significantly higher than that extracted by the Epicentre kit (t-test, p-value=0.0425). There was no significant difference (Kruskal–Wallis test, p-value=0.682) in the observed species richness between the samples extracted by both methods. However, the sequencing depth did not completely capture the whole OTU richness in the kit samples, since, with a higher number of sequences in the chemical protocol samples, the curve was still rising and did not reach a plateau in the “observed species curve”. The importance of adequate sequencing depth for obtaining an accurate estimation of the diversity in a sample was studied previously, and it was indicated that an increased sequencing depth markedly improves the estimates of diversity.19
There was no significant difference in both the within community diversity (alpha diversity) and the between community diversity (beta diversity) between the samples extracted by both methods. All these results indicate a greater efficiency of the developed chemical protocol to yield high-quality metagenomic DNA comparable to that extracted by the well-established Epicentre kit.
When the tentative cost estimation of the optimized method was attempted, it was found to be approximately 0.125 USD for processing one sample (Supplementary Table S1); this may be attributed to the use of low-cost materials. The purifying agents used in the developed protocol are relatively inexpensive compared to other purifying agents as anion resins and hydroxyapatite columns. In comparison with any other commercially available kit, the cost of the high-quality DNA isolation from environmental water sample using the developed chemical protocol is remarkably low (processing one sample using Epicentre kit costs about 6.85 USD). On the other hand, the time required for metagenomic DNA extraction did not vary much for either of the studied protocols; Epicentre kit and the developed chemical protocol required 1.5h and 2h, respectively, for metagenomic DNA isolation.
In conclusion, a cost and time-efficient chemical protocol for the extraction of mDNA from environmental water samples has been developed. The efficient recovery and high quality of the extracted DNA using the developed chemical protocol makes it compatible with high-resolution molecular applications including next-generation sequencing. This proposes the developed chemical protocol to be a gold standard in future metagenomic studies investigating a large number of samples.
Authors’ contributionsMariam Hassan and Tamer Essam conceived and designed the study. Mariam Hassan performed the experiments. Mariam Hassan and Tamer Essam analyzed the data. Mariam Hassan prepared the figures and illustrations. Mariam Hassan, Tamer Essam, and Salwa Megahed drafted the manuscript. Mariam Hassan and Tamer Essam wrote the paper in final format. All authors read and approved the final version of the manuscript.
FundingThis work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflicts of interestAll authors declare that they have no conflict of interest.
The authors are deeply grateful to Dr. Alex Mira, Department of Genomics and Health, Center for Advanced Research in Public Health (CSISP), Valencia, Spain, for sequencing the samples.
The following are the supplementary data to this article: